Department of Health Sciences, University of Leicester, Leicester, UK.
Division of Pulmonary and Critical Care Medicine, University of Michigan, Ann Arbor, MI, USA.
Lancet Respir Med. 2023 Jan;11(1):65-73. doi: 10.1016/S2213-2600(22)00251-X. Epub 2022 Aug 16.
Idiopathic pulmonary fibrosis (IPF) is an incurable lung disease characterised by progressive scarring leading to alveolar stiffness, reduced lung capacity, and impeded gas transfer. We aimed to identify genetic variants associated with declining lung capacity or declining gas transfer after diagnosis of IPF.
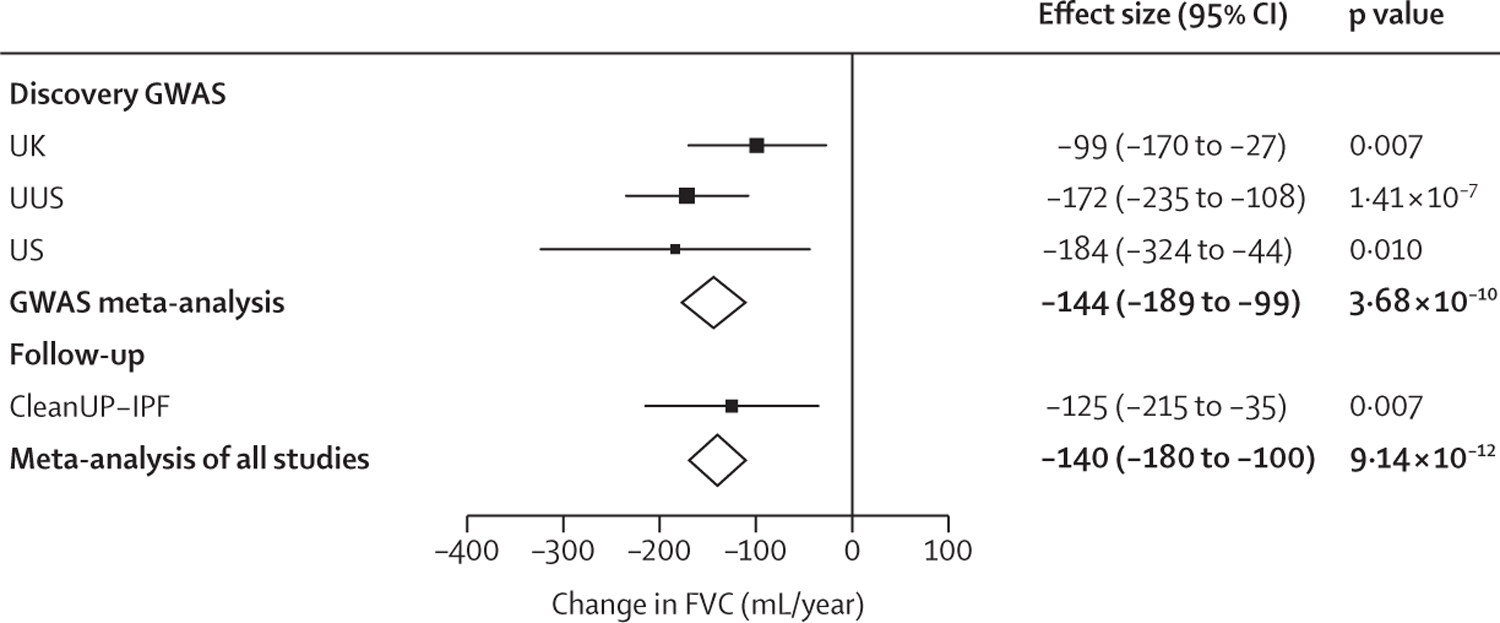
We did a genome-wide meta-analysis of longitudinal measures of forced vital capacity (FVC) and diffusing capacity of the lung for carbon monoxide (DLCO) in individuals diagnosed with IPF. Individuals were recruited to three studies between June, 1996, and August, 2017, from across centres in the US, UK, and Spain. Suggestively significant variants were investigated further in an additional independent study (CleanUP-IPF). All four studies diagnosed cases following American Thoracic Society/European Respiratory Society guidelines. Variants were defined as significantly associated if they had a meta-analysis p<5 × 10 when meta-analysing across all discovery and follow-up studies, had consistent direction of effects across all four studies, and were nominally significant (p<0·05) in each study.
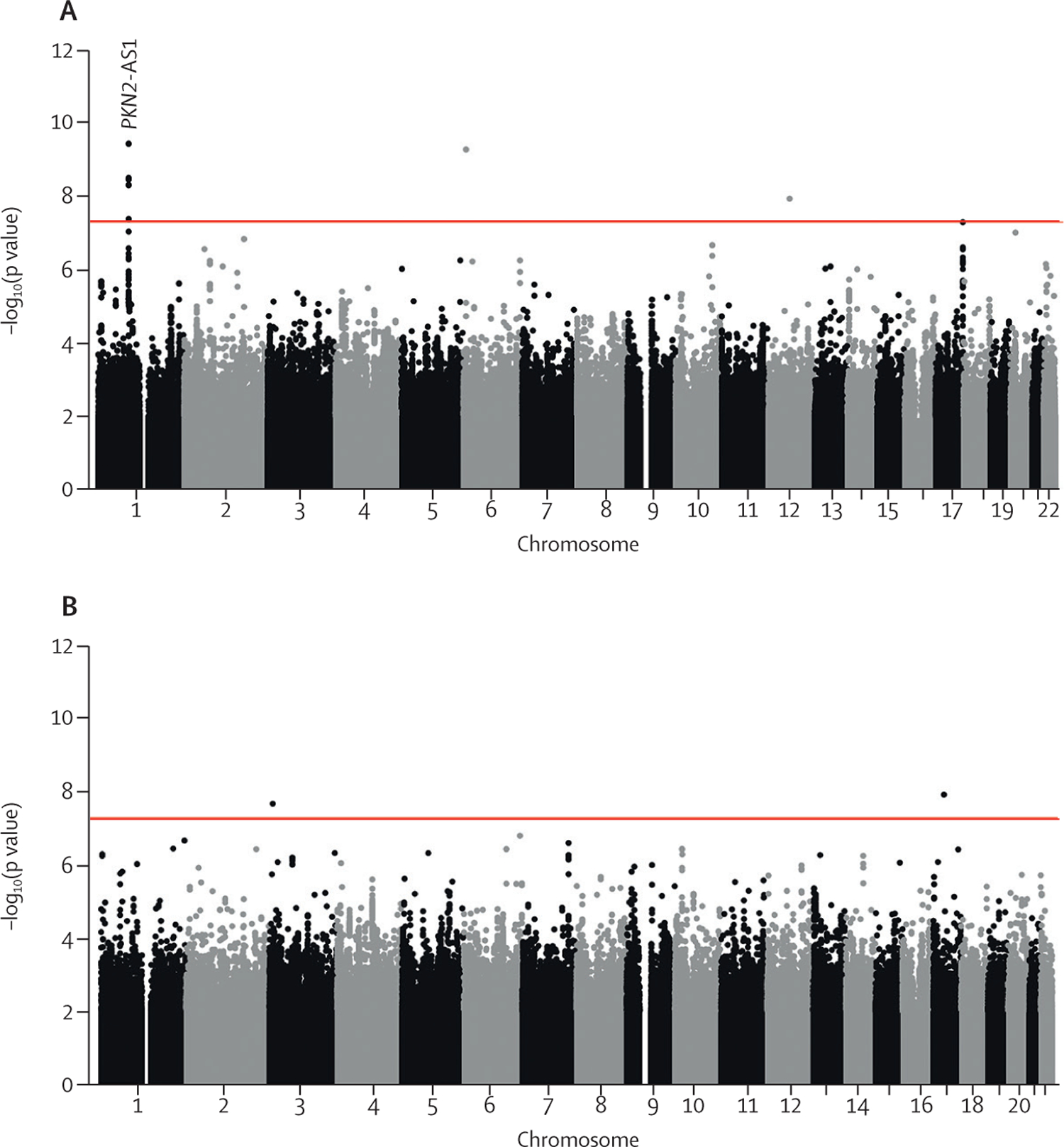
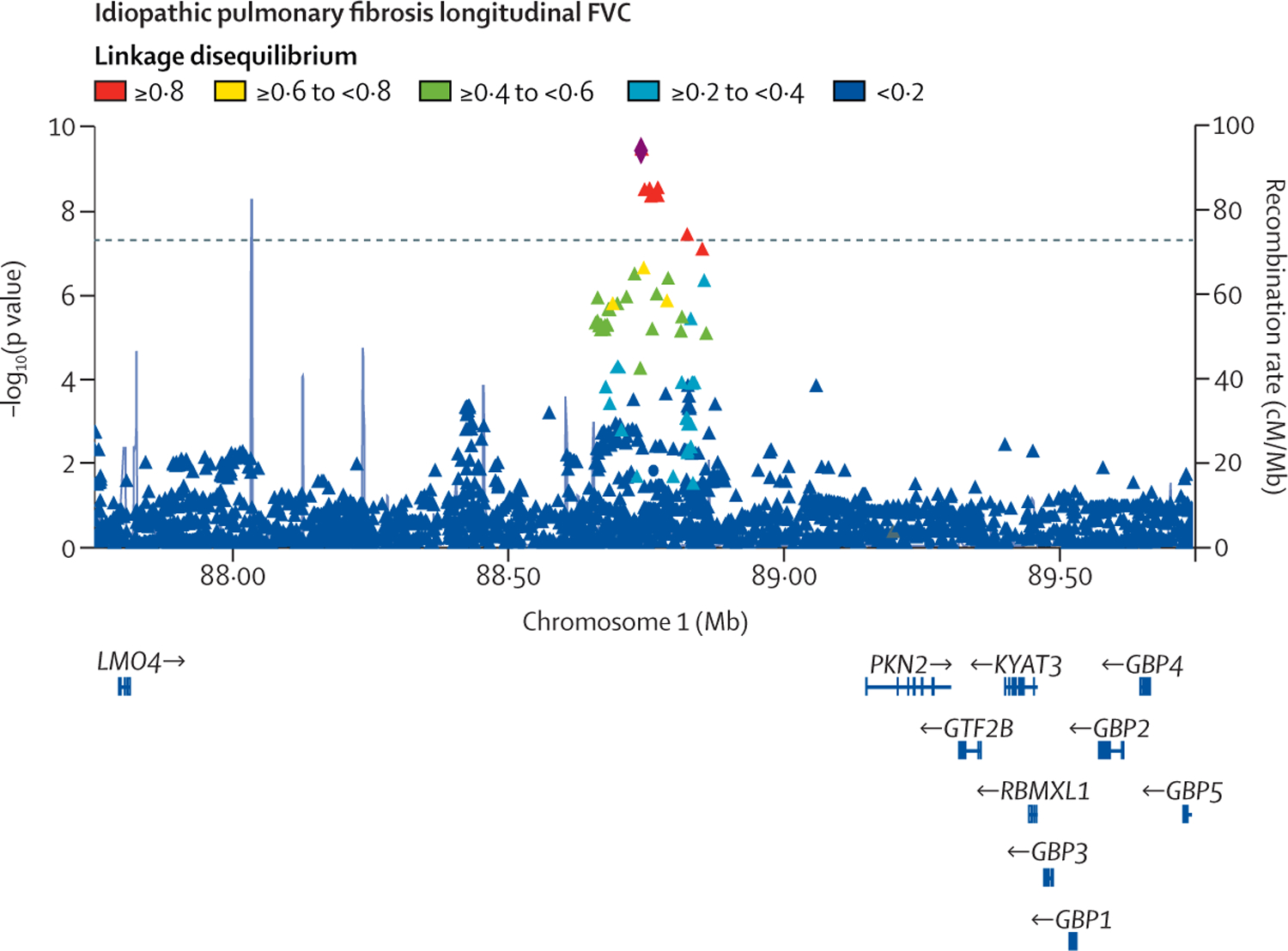
1329 individuals with a total of 5216 measures were included in the FVC analysis. 975 individuals with a total of 3361 measures were included in the DLCO analysis. For the discovery genome-wide analyses, 7 611 174 genetic variants were included in the FVC analysis and 7 536 843 in the DLCO analysis. One variant (rs115982800) located in an antisense RNA gene for protein kinase N2 (PKN2) showed a genome-wide significant association with FVC decline (-140 mL/year per risk allele [95% CI -180 to -100]; p=9·14 × 10).
Our analysis identifies a genetic variant associated with disease progression, which might highlight a new biological mechanism for IPF. We found that PKN2, a Rho and Rac effector protein, is the most likely gene of interest from this analysis. PKN2 inhibitors are currently in development and signify a potential novel therapeutic approach for IPF.
Action for Pulmonary Fibrosis, Medical Research Council, Wellcome Trust, and National Institutes of Health National Heart, Lung, and Blood Institute.
特发性肺纤维化(IPF)是一种无法治愈的肺部疾病,其特征是进行性瘢痕形成导致肺泡僵硬、肺容量减少和气体转移受阻。我们旨在确定与 IPF 诊断后肺容量下降或气体转移下降相关的遗传变异。
我们对诊断为 IPF 的个体的用力肺活量(FVC)和一氧化碳弥散量(DLCO)的纵向测量值进行了全基因组荟萃分析。这些个体于 1996 年 6 月至 2017 年 8 月期间从美国、英国和西班牙的各个中心招募到三个研究中。在一个额外的独立研究(CleanUP-IPF)中进一步研究了提示性显著的变异。所有四项研究均按照美国胸科学会/欧洲呼吸学会的指南诊断病例。如果在整个发现和随访研究中进行荟萃分析时变异具有 5×10-5以下的 meta 分析 p 值、在所有四项研究中具有一致的效应方向并且在每个研究中具有名义上的显著意义(p<0.05),则将其定义为显著相关。
共有 1329 名个体进行了 5216 次 FVC 分析,共有 975 名个体进行了 3361 次 DLCO 分析。对于发现全基因组分析,FVC 分析中包含 7611174 个遗传变异,DLCO 分析中包含 7536843 个遗传变异。一个位于蛋白激酶 N2(PKN2)反义 RNA 基因中的变异(rs115982800)与 FVC 下降呈全基因组显著相关(每风险等位基因下降 140 毫升/年[95%CI-180 至-100];p=9.14×10)。
我们的分析确定了与疾病进展相关的遗传变异,这可能突出了 IPF 的新生物学机制。我们发现,PKN2,一种 Rho 和 Rac 效应蛋白,是该分析中最有可能的感兴趣基因。PKN2 抑制剂目前正在开发中,代表了 IPF 的一种潜在新治疗方法。
肺纤维化行动、英国医学研究理事会、惠康信托基金会和美国国立卫生研究院国家心肺血液研究所。