The African Computational Genomics (TACG) Research Group, Medical Research Council/Uganda Virus Research Institute and London School of Hygiene & Tropical Medicine Uganda Research Unit, Entebbe 31405, Uganda.
Department of Immunology and Molecular Biology, College of Health Sciences, Makerere University, Kampala 10101, Uganda.
Genes (Basel). 2022 Aug 16;13(8):1460. doi: 10.3390/genes13081460.
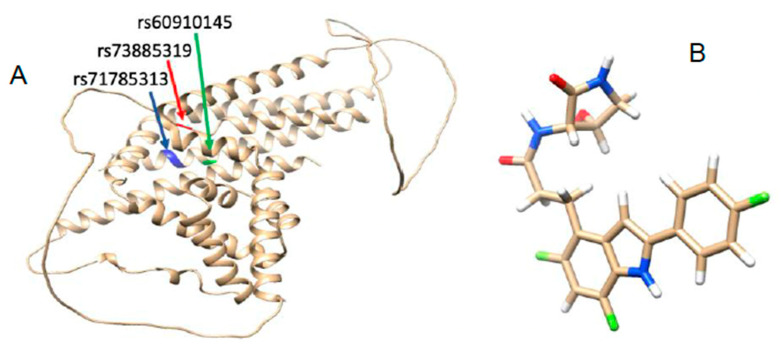
According to observational studies, two polymorphisms in the apolipoprotein L1 () gene have been linked to an increased risk of chronic kidney disease (CKD) in Africans. One polymorphism involves the substitution of two amino-acid residues (S342G and I384M; known as G1), while the other involves the deletion of two amino-acid residues in a row (N388 and Y389; termed G2). Despite the strong link between polymorphisms and kidney disease, the molecular mechanisms via which these mutations influence the onset and progression of CKD remain unknown.
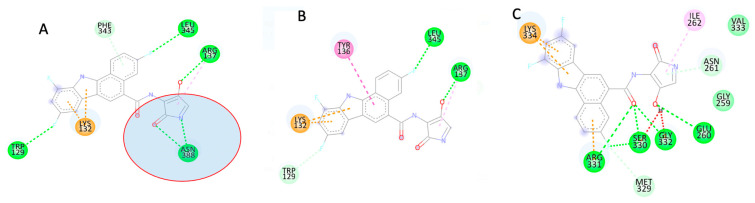
To predict the active site and allosteric site on the protein, we used the Computed Atlas of Surface Topography of Proteins (CASTp) and the Protein Allosteric Sites Server (PASSer). Using an extended molecular dynamics simulation, we investigated the characteristic structural perturbations in the 3D structures of variants.
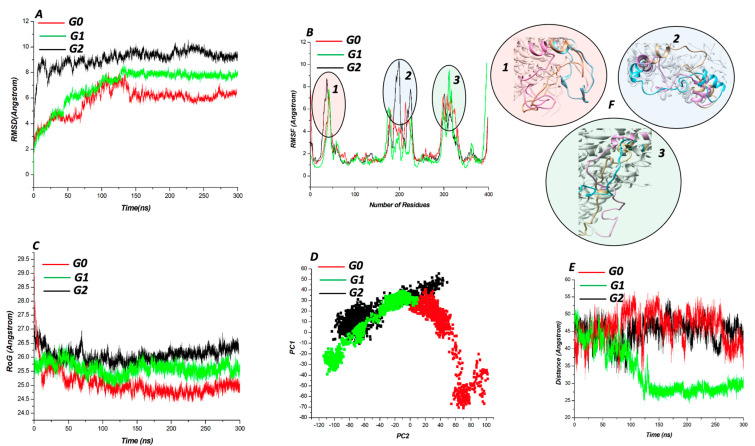
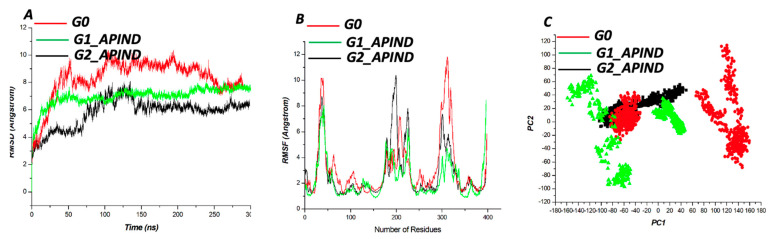
According to CASTp's active site characterization, the topmost predicted site had a surface area of 964.892 Å and a pocket volume of 900.792 Å. For the top three allosteric pockets, the allostery probability was 52.44%, 46.30%, and 38.50%, respectively. The systems reached equilibrium in about 125 ns. From 0-100 ns, there was also significant structural instability. When compared to G1 and G2, the wildtype protein (G0) had overall high stability throughout the simulation. The root-mean-square fluctuation (RMSF) of wildtype and variant protein backbone Cα fluctuations revealed that the Cα of the variants had a large structural fluctuation when compared to the wildtype.
Using a combination of different computational techniques, we identified binding sites within the protein that could be an attractive site for potential inhibitors of . Furthermore, the G1 and G2 mutations reduced the structural stability of .
根据观察性研究,载脂蛋白 L1()基因中的两个多态性与非洲人慢性肾脏病(CKD)的风险增加有关。一个多态性涉及两个氨基酸残基的取代(S342G 和 I384M;称为 G1),另一个多态性涉及两个氨基酸残基的连续缺失(N388 和 Y389;称为 G2)。尽管与多态性和肾脏疾病之间存在很强的联系,但这些突变影响 CKD 发病和进展的分子机制仍不清楚。
为了预测蛋白上的活性位点和别构位点,我们使用了计算蛋白质表面拓扑图谱(CASTp)和蛋白别构位点服务器(PASSer)。通过扩展分子动力学模拟,我们研究了变体 3D 结构中特征结构扰动。
根据 CASTp 的活性位点特征化,预测的最上面的位点表面积为 964.892 Å,口袋体积为 900.792 Å。对于前三个别构口袋,别构概率分别为 52.44%、46.30%和 38.50%。系统在大约 125 ns 达到平衡。在 0-100 ns 之间,也存在显著的结构不稳定性。与 G1 和 G2 相比,野生型蛋白(G0)在整个模拟过程中具有整体高稳定性。野生型和变体蛋白骨架 Cα波动的均方根波动(RMSF)表明,与野生型相比,变体的 Cα具有较大的结构波动。
使用多种计算技术的组合,我们确定了蛋白内的结合位点,这些位点可能是潜在抑制剂的有吸引力的靶点。此外,G1 和 G2 突变降低了变体的结构稳定性。