Department of Pharmacology, College of Medicine, China Medical University, Taichung 40402, Taiwan.
Ph.D. Program for Biotech Pharmaceutical Industry, China Medical University, Taichung 40402, Taiwan.
Int J Mol Sci. 2022 Aug 13;23(16):9086. doi: 10.3390/ijms23169086.
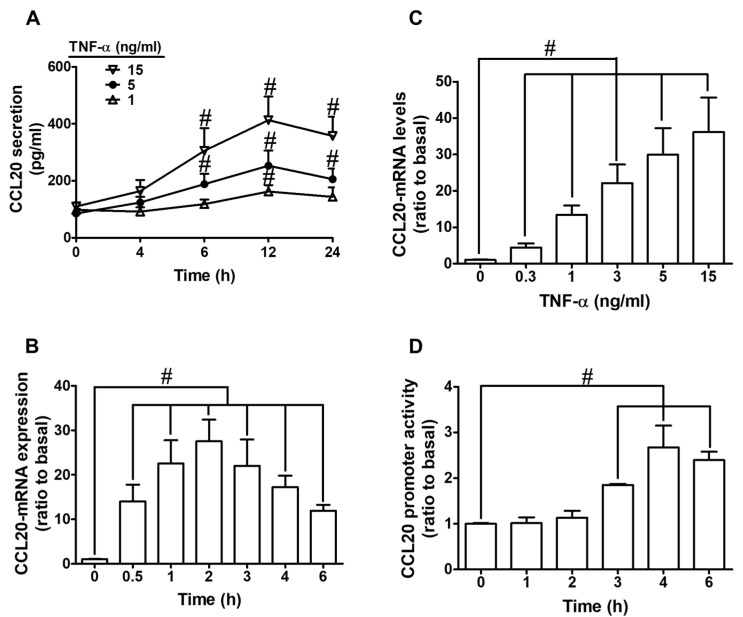
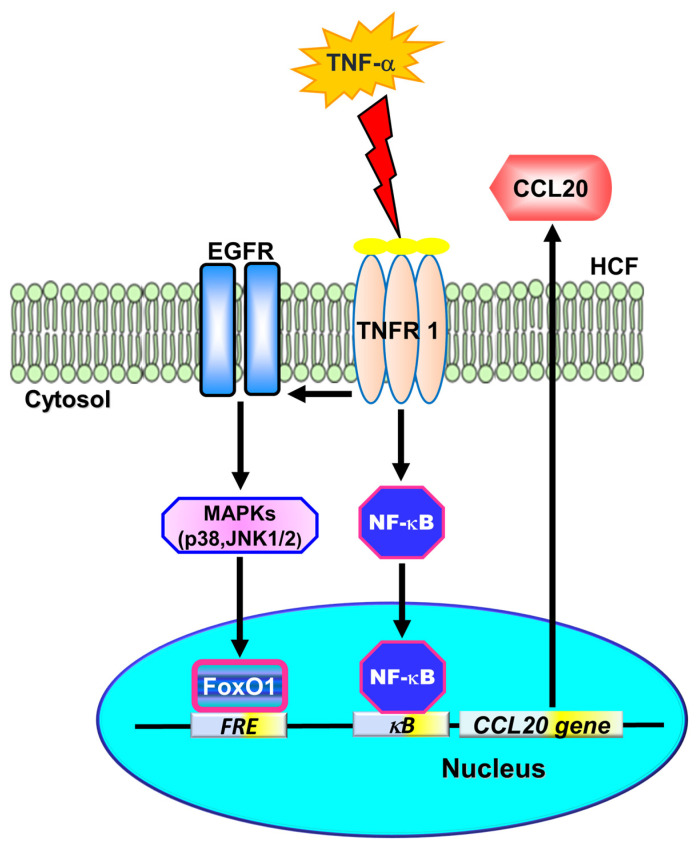
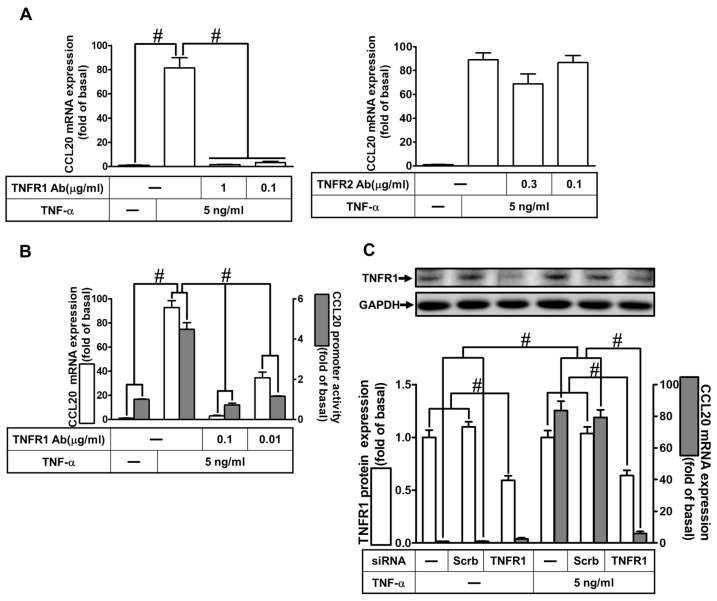
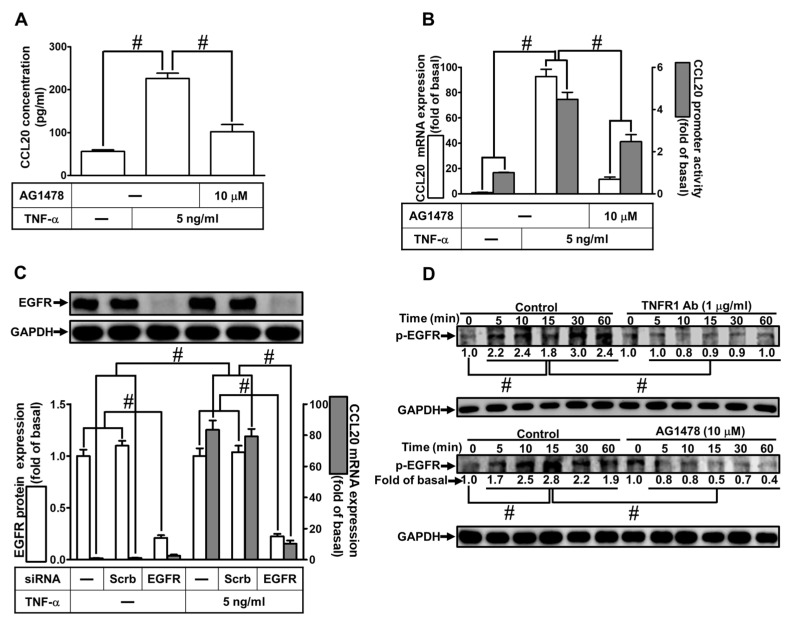
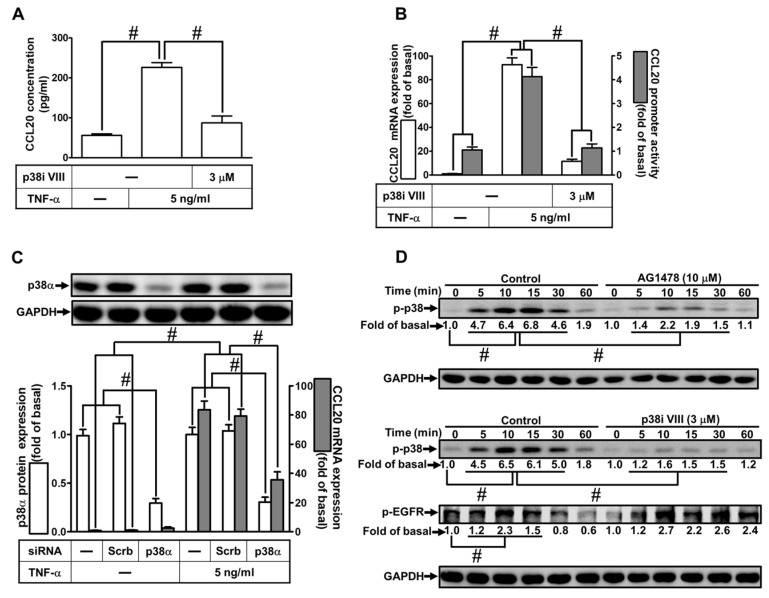
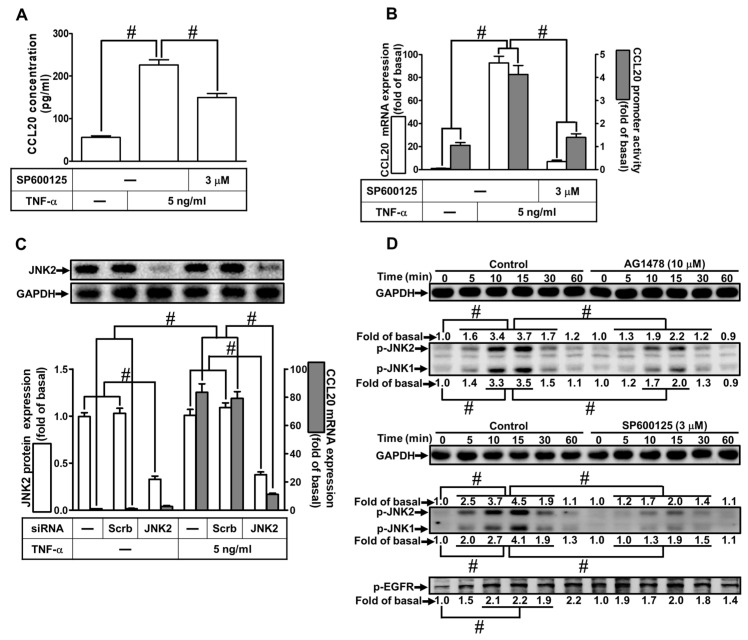
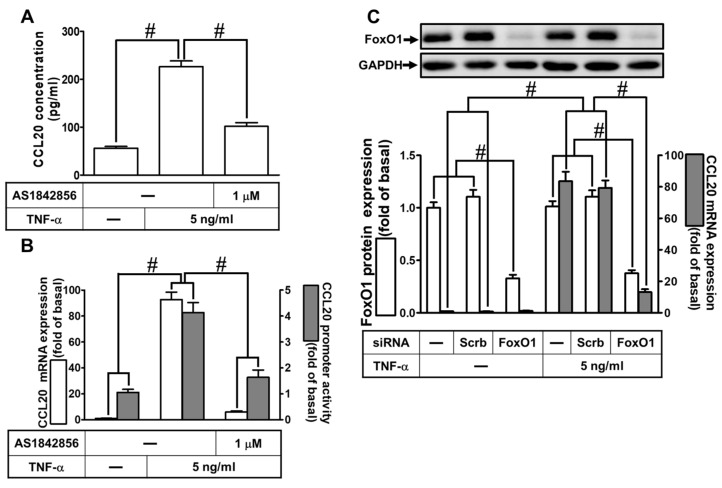
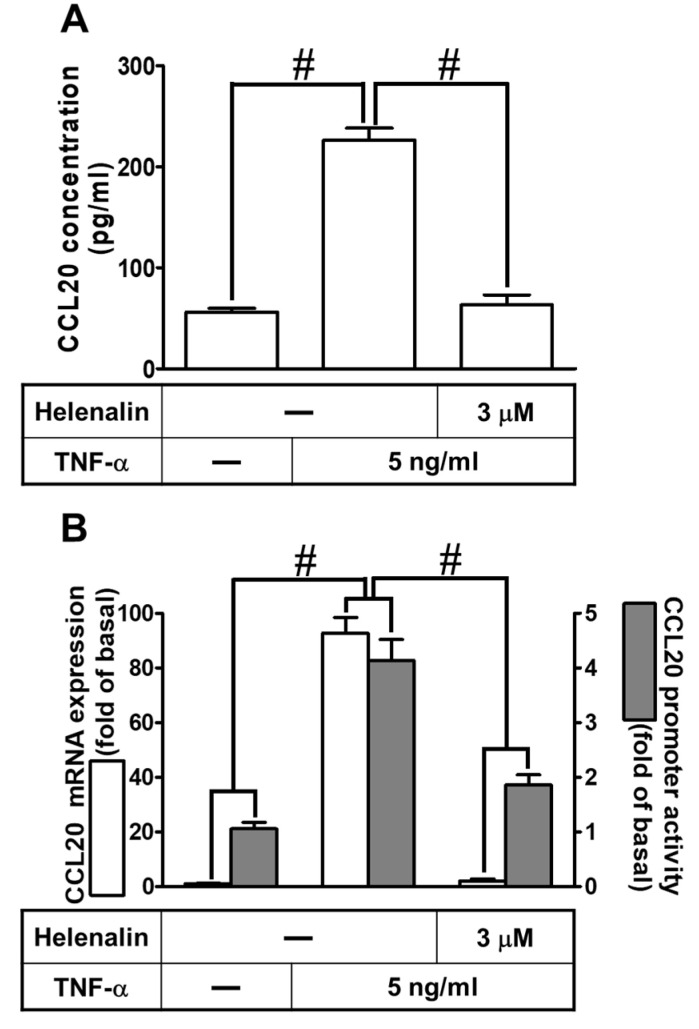
Tumor necrosis factor (TNF)-α is involved in the pathogenesis of cardiac injury, inflammation, and apoptosis. It is a crucial pro-inflammatory cytokine in many heart disorders, including chronic heart failure and ischemic heart disease, contributing to cardiac remodeling and dysfunction. The implication of TNF-α in inflammatory responses in the heart has been indicated to be mediated through the induction of C-C Motif Chemokine Ligand 20 (CCL20). However, the detailed mechanisms of TNF-α-induced CCL20 upregulation in human cardiac fibroblasts (HCFs) are not completely defined. We demonstrated that in HCFs, TNF-α induced CCL20 mRNA expression and promoter activity leading to an increase in the secretion of CCL20. TNF-α-mediated responses were attenuated by pretreatment with TNFR1 antibody, the inhibitor of epidermal growth factor receptor (EGFR) (AG1478), p38 mitogen-activated protein kinase (MAPK) (p38 inhibitor VIII, p38i VIII), c-Jun amino N-terminal kinase (JNK)1/2 (SP600125), nuclear factor kappaB (NF-κB) (helenalin), or forkhead box O (FoxO)1 (AS1841856) and transfection with siRNA of TNFR1, EGFR, p38α, JNK2, p65, or FoxO1. Moreover, TNF-α markedly induced EGFR, p38 MAPK, JNK1/2, FoxO1, and NF-κB p65 phosphorylation which was inhibited by their respective inhibitors in these cells. In addition, TNF-α-enhanced binding of FoxO1 or p65 to the CCL20 promoter was inhibited by p38i VIII, SP600125, and AS1841856, or helenalin, respectively. Accordingly, in HCFs, our findings are the first to clarify that TNF-α-induced CCL20 secretion is mediated through a TNFR1-dependent EGFR/p38 MAPK and JNK1/2/FoxO1 or NF-κB cascade. We demonstrated that TNFR1-derived EGFR transactivation is involved in the TNF-α-induced responses in these cells. Understanding the regulation of CCL20 expression by TNF-α on HCFs may provide a potential therapeutic strategy in cardiac inflammatory disorders.
肿瘤坏死因子 (TNF)-α 参与心脏损伤、炎症和细胞凋亡的发病机制。它是许多心脏疾病(包括慢性心力衰竭和缺血性心脏病)中重要的促炎细胞因子,导致心脏重构和功能障碍。TNF-α 在心脏炎症反应中的作用已被证明是通过诱导 C-C 基序趋化因子配体 20(CCL20)来介导的。然而,TNF-α 在人心脏成纤维细胞(HCFs)中诱导 CCL20 上调的详细机制尚不完全明确。我们证明在 HCFs 中,TNF-α 诱导 CCL20 mRNA 表达和启动子活性,导致 CCL20 的分泌增加。TNF-α 介导的反应可被 TNFR1 抗体、表皮生长因子受体(EGFR)抑制剂(AG1478)、p38 丝裂原活化蛋白激酶(MAPK)(p38 抑制剂 VIII,p38i VIII)、c-Jun 氨基末端激酶(JNK)1/2(SP600125)、核因子 kappaB(NF-κB)(helenalin)或叉头框 O(FoxO)1(AS1841856)预处理以及用 TNFR1、EGFR、p38α、JNK2、p65 或 FoxO1 的 siRNA 转染来减弱。此外,TNF-α 明显诱导 EGFR、p38 MAPK、JNK1/2、FoxO1 和 NF-κB p65 在这些细胞中的磷酸化,而这些磷酸化可被其各自的抑制剂抑制。此外,TNF-α 增强的 FoxO1 或 p65 与 CCL20 启动子的结合可被 p38i VIII、SP600125 和 AS1841856 或 helenalin 分别抑制。因此,在 HCFs 中,我们的研究结果首次阐明 TNF-α 诱导的 CCL20 分泌是通过 TNFR1 依赖性 EGFR/p38 MAPK 和 JNK1/2/FoxO1 或 NF-κB 级联来介导的。我们证明了 TNFR1 衍生的 EGFR 转激活参与了这些细胞中 TNF-α 诱导的反应。了解 TNF-α 对 HCFs 中 CCL20 表达的调节可能为心脏炎症性疾病提供一种潜在的治疗策略。