Cele Nosipho, Awolade Paul, Seboletswe Pule, Olofinsan Kolawole, Islam Md Shahidul, Singh Parvesh
School of Chemistry and Physics, University of KwaZulu-Natal, P/Bag X54001, Westville, Durban 4000, South Africa.
Department of Biochemistry, School of Life Sciences, University of Kwazulu-Natal, Westville, Durban 4000, South Africa.
Pharmaceuticals (Basel). 2022 Aug 22;15(8):1035. doi: 10.3390/ph15081035.
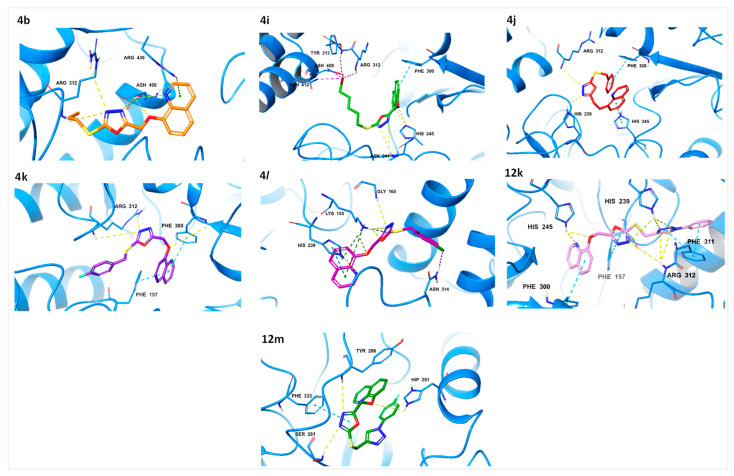
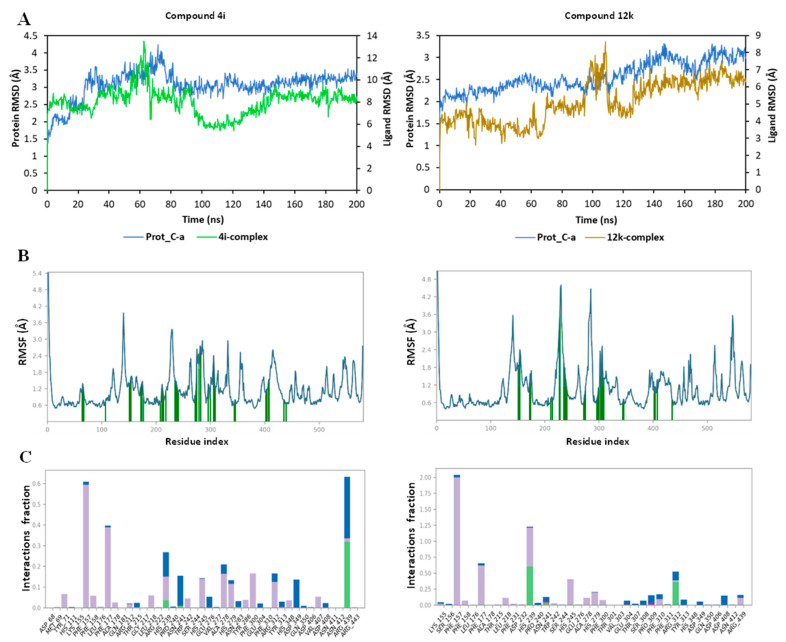
Diabetes mellitus (DM) is a multifaceted metabolic disorder that remains a major threat to global health security. Sadly, the clinical relevance of available drugs is burdened with an upsurge in adverse effects; hence, inhibiting the carbohydrate-hydrolyzing enzymes α-glucosidase and α-amylase while preventing oxidative stress is deemed a practicable strategy for regulating postprandial glucose levels in DM patients. We report herein the α-glucosidase and α-amylase inhibition and antioxidant profile of quinoline hybrids - and - bearing 1,3,4-oxadiazole and 1,2,3-triazole cores, respectively. Overall, compound with a bromopentyl sidechain exhibited the strongest α-glucosidase inhibition (IC = 15.85 µM) relative to reference drug acarbose (IC = 17.85 µM) and the best antioxidant profile in FRAP, DPPH, and NO scavenging assays. Compounds and also emerged as the most potent NO scavengers (IC = 2.67 and 3.01 µM, respectively) compared to gallic acid (IC = 728.68 µM), while notable α-glucosidase inhibition was observed for -fluorobenzyl compound (IC = 23.69 µM) and phenyl-1,2,3-triazolyl compound (IC = 22.47 µM). Moreover, kinetic studies established the mode of α-glucosidase inhibition as non-competitive, thus classifying the quinoline hybrids as allosteric inhibitors. Molecular docking and molecular dynamics simulations then provided insights into the protein-ligand interaction profile and the stable complexation of promising hybrids at the allosteric site of α-glucosidase. These results showcase these compounds as worthy scaffolds for developing more potent α-glucosidase inhibitors with antioxidant activity for effective DM management.
糖尿病(DM)是一种多方面的代谢紊乱疾病,仍然是全球健康安全的重大威胁。遗憾的是,现有药物的临床相关性因不良反应激增而受到影响;因此,抑制碳水化合物水解酶α-葡萄糖苷酶和α-淀粉酶同时预防氧化应激被认为是调节糖尿病患者餐后血糖水平的可行策略。我们在此报告分别带有1,3,4-恶二唑和1,2,3-三唑核心的喹啉杂化物对α-葡萄糖苷酶和α-淀粉酶的抑制作用以及抗氧化特性。总体而言,带有溴戊基侧链的化合物相对于参考药物阿卡波糖(IC = 17.85 µM)表现出最强的α-葡萄糖苷酶抑制作用(IC = 15.85 µM)以及在铁离子还原抗氧化能力(FRAP)、二苯基苦味酰基自由基(DPPH)和一氧化氮(NO)清除试验中最佳的抗氧化特性。与没食子酸(IC = 728.68 µM)相比,化合物 和 也成为最有效的NO清除剂(分别为IC = 2.67 和3.01 µM),而对于-氟苄基化合物 (IC = 23.69 µM)和苯基-1,2,3-三唑基化合物 (IC = 22.47 µM)观察到显著的α-葡萄糖苷酶抑制作用。此外动力学研究确定α-葡萄糖苷酶的抑制模式为非竞争性,从而将喹啉杂化物归类为变构抑制剂。分子对接和分子动力学模拟随后提供了有关蛋白质-配体相互作用特征以及有前景的杂化物在α-葡萄糖苷酶变构位点的稳定络合的见解。这些结果表明这些化合物是用于开发更有效的具有抗氧化活性以有效管理糖尿病的α-葡萄糖苷酶抑制剂的有价值骨架。