Lepamets Maarja, Auwerx Chiara, Nõukas Margit, Claringbould Annique, Porcu Eleonora, Kals Mart, Jürgenson Tuuli, Morris Andrew Paul, Võsa Urmo, Bochud Murielle, Stringhini Silvia, Wijmenga Cisca, Franke Lude, Peterson Hedi, Vilo Jaak, Lepik Kaido, Mägi Reedik, Kutalik Zoltán
Estonian Genome Centre, Institute of Genomics, University of Tartu, Tartu 51010, Estonia.
Institute of Molecular and Cell Biology, University of Tartu, Tartu 51010, Estonia.
HGG Adv. 2022 Aug 1;3(4):100133. doi: 10.1016/j.xhgg.2022.100133. eCollection 2022 Oct 13.
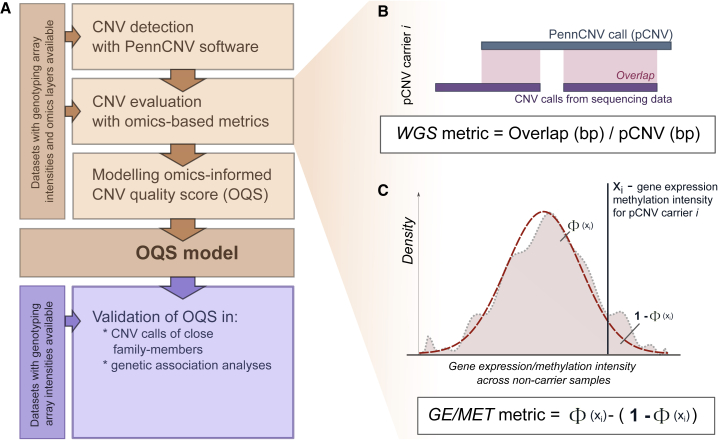
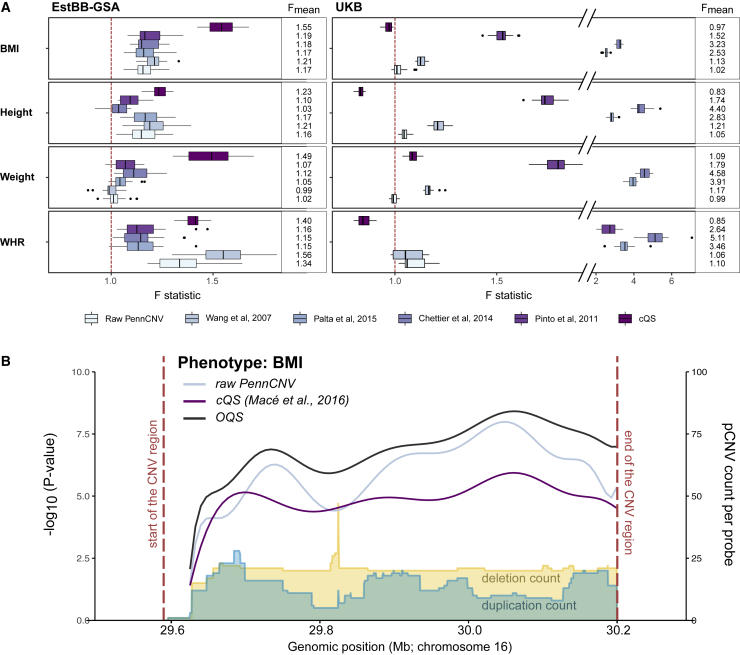
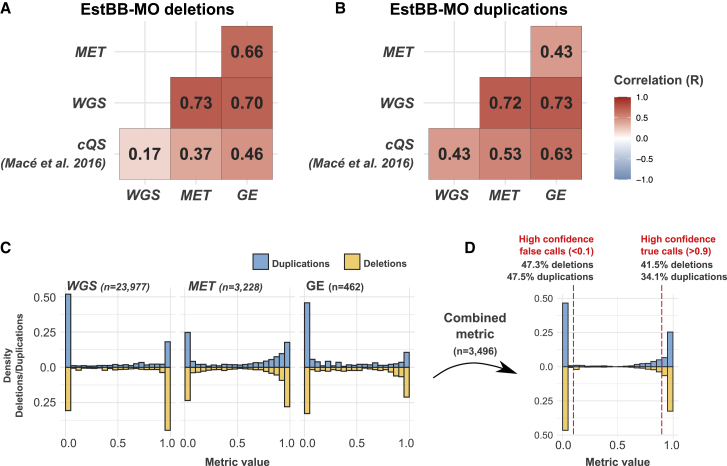
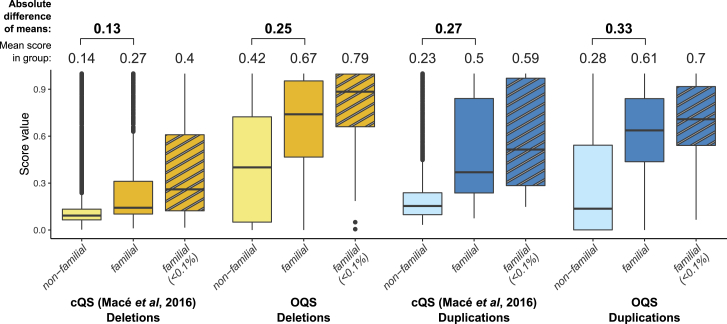
Copy-number variations (CNV) are believed to play an important role in a wide range of complex traits, but discovering such associations remains challenging. While whole-genome sequencing (WGS) is the gold-standard approach for CNV detection, there are several orders of magnitude more samples with available genotyping microarray data. Such array data can be exploited for CNV detection using dedicated software (e.g., PennCNV); however, these calls suffer from elevated false-positive and -negative rates. In this study, we developed a CNV quality score that weights PennCNV calls (pCNVs) based on their likelihood of being true positive. First, we established a measure of pCNV reliability by leveraging evidence from multiple omics data (WGS, transcriptomics, and methylomics) obtained from the same samples. Next, we built a predictor of omics-confirmed pCNVs, termed omics-informed quality score (OQS), using only PennCNV software output parameters. Promisingly, OQS assigned to pCNVs detected in close family members was up to 35% higher than the OQS of pCNVs not carried by other relatives (p < 3.0 × 10), outperforming other scores. Finally, in an association study of four anthropometric traits in 89,516 Estonian Biobank samples, the use of OQS led to a relative increase in the trait variance explained by CNVs of up to 56% compared with published quality filtering methods or scores. Overall, we put forward a flexible framework to improve any CNV detection method leveraging multi-omics evidence, applied it to improve PennCNV calls, and demonstrated its utility by improving the statistical power for downstream association analyses.
拷贝数变异(CNV)被认为在广泛的复杂性状中起重要作用,但发现此类关联仍然具有挑战性。虽然全基因组测序(WGS)是CNV检测的金标准方法,但有可用基因分型微阵列数据的样本数量要多几个数量级。此类阵列数据可使用专用软件(如PennCNV)用于CNV检测;然而,这些调用存在较高的假阳性和假阴性率。在本研究中,我们开发了一种CNV质量评分,根据其为真阳性的可能性对PennCNV调用(pCNV)进行加权。首先,我们通过利用从相同样本获得的多种组学数据(WGS、转录组学和甲基组学)的证据,建立了一种pCNV可靠性的度量。接下来,我们仅使用PennCNV软件输出参数构建了一个经组学确认的pCNV预测器,称为组学知情质量评分(OQS)。很有前景的是,分配给在近亲中检测到的pCNV的OQS比其他亲属未携带的pCNV的OQS高出多达35%(p < 3.0×10),优于其他评分。最后,在对89516个爱沙尼亚生物银行样本的四项人体测量性状的关联研究中,与已发表的质量过滤方法或评分相比,使用OQS导致CNV解释的性状方差相对增加高达56%。总体而言,我们提出了一个灵活的框架,以利用多组学证据改进任何CNV检测方法,将其应用于改进PennCNV调用,并通过提高下游关联分析的统计效力证明了其效用。