Sorbonne Université, CNRS, UMR 7238, Laboratoire de Biologie Computationnelle et Quantitative, Paris, France.
Sorbonne Université, AP-HP, Hôpital Pitié-Salpêtrière, UMR_S 1138 Department of Hematology, Paris, France.
PLoS Comput Biol. 2022 Aug 29;18(8):e1010411. doi: 10.1371/journal.pcbi.1010411. eCollection 2022 Aug.
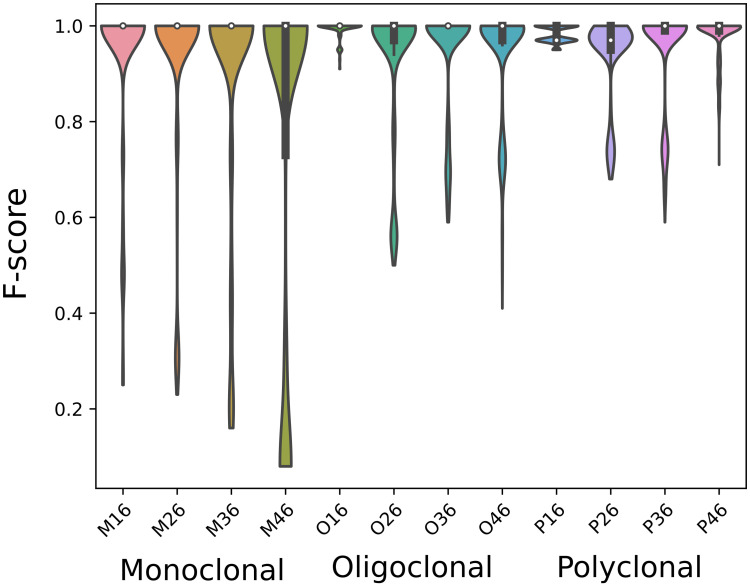
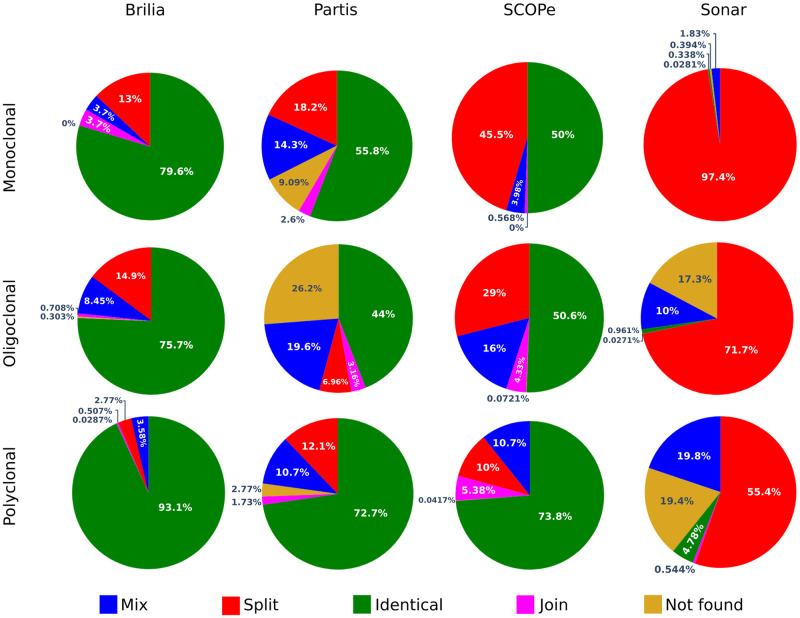
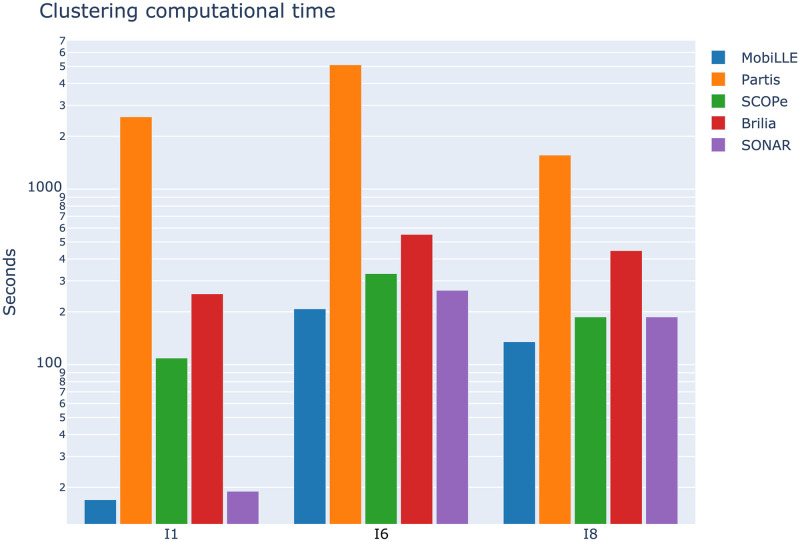
The adaptive B cell response is driven by the expansion, somatic hypermutation, and selection of B cell clonal lineages. A high number of clonal lineages in a B cell population indicates a highly diverse repertoire, while clonal size distribution and sequence diversity reflect antigen selective pressure. Identifying clonal lineages is fundamental to many repertoire studies, including repertoire comparisons, clonal tracking, and statistical analysis. Several methods have been developed to group sequences from high-throughput B cell repertoire data. Current methods use clustering algorithms to group clonally-related sequences based on their similarities or distances. Such approaches create groups by optimizing a single objective that typically minimizes intra-clonal distances. However, optimizing several objective functions can be advantageous and boost the algorithm convergence rate. Here we propose MobiLLe, a new method based on multi-objective clustering. Our approach requires V(D)J annotations to obtain the initial groups and iteratively applies two objective functions that optimize cohesion and separation within clonal lineages simultaneously. We show that our method greatly improves clonal lineage grouping on simulated benchmarks with varied mutation rates compared to other tools. When applied to experimental repertoires generated from high-throughput sequencing, its clustering results are comparable to the most performing tools and can reproduce the results of previous publications. The method based on multi-objective clustering can accurately identify clonally-related antibody sequences and presents the lowest running time among state-of-art tools. All these features constitute an attractive option for repertoire analysis, particularly in the clinical context. MobiLLe can potentially help unravel the mechanisms involved in developing and evolving B cell malignancies.
适应性 B 细胞反应是由 B 细胞克隆谱系的扩增、体细胞超突变和选择驱动的。B 细胞群体中大量的克隆谱系表明其多样性较高,而克隆大小分布和序列多样性反映了抗原的选择压力。鉴定克隆谱系是许多免疫库研究的基础,包括免疫库比较、克隆追踪和统计分析。已经开发了几种方法来对高通量 B 细胞免疫库数据中的序列进行分组。目前的方法使用聚类算法根据序列之间的相似性或距离将克隆相关的序列分组。这些方法通过优化单个目标(通常是最小化克隆内的距离)来创建组。然而,优化多个目标函数可能会更有利,并且可以提高算法的收敛速度。在这里,我们提出了 MobiLLe,这是一种基于多目标聚类的新方法。我们的方法需要 V(D)J 注释来获得初始组,然后迭代地应用两个目标函数,同时优化克隆谱系内的凝聚和分离。我们表明,与其他工具相比,我们的方法在具有不同突变率的模拟基准上大大提高了克隆谱系分组的性能。当应用于高通量测序产生的实验免疫库时,其聚类结果与性能最好的工具相当,并可以重现以前出版物的结果。基于多目标聚类的方法可以准确识别克隆相关的抗体序列,并且在最先进的工具中运行时间最短。所有这些功能构成了分析免疫库的一个有吸引力的选择,特别是在临床环境中。MobiLLe 有可能帮助揭示 B 细胞恶性肿瘤发展和演变的机制。