Lee Donald W, Khavrutskii Ilja V, Wallqvist Anders, Bavari Sina, Cooper Christopher L, Chaudhury Sidhartha
Biotechnology HPC Software Applications Institute (BHSAI), Telemedicine and Advanced Technology Research Center, U.S. Army Medical Research and Materiel Command , Fort Detrick, MD , USA.
Molecular and Translational Sciences, U.S. Army Medical Research Institute of Infectious Diseases , Frederick, MD , USA.
Front Immunol. 2017 Jan 17;7:681. doi: 10.3389/fimmu.2016.00681. eCollection 2016.
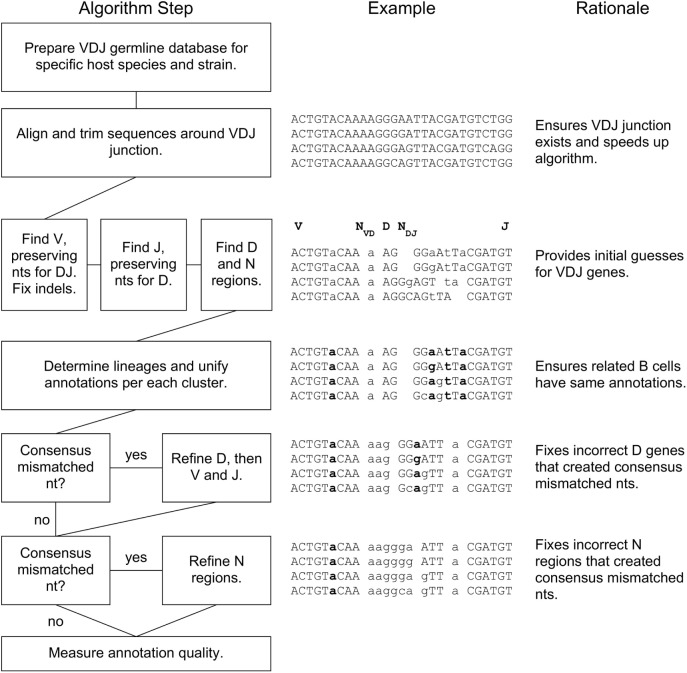
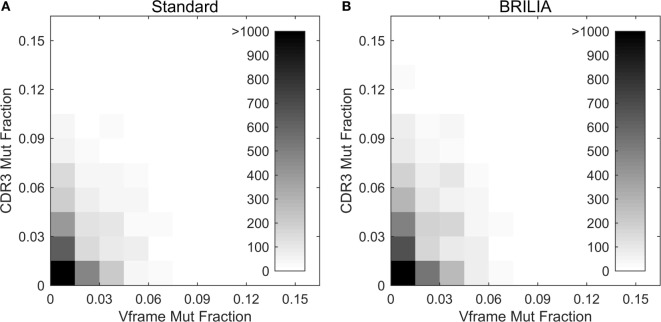
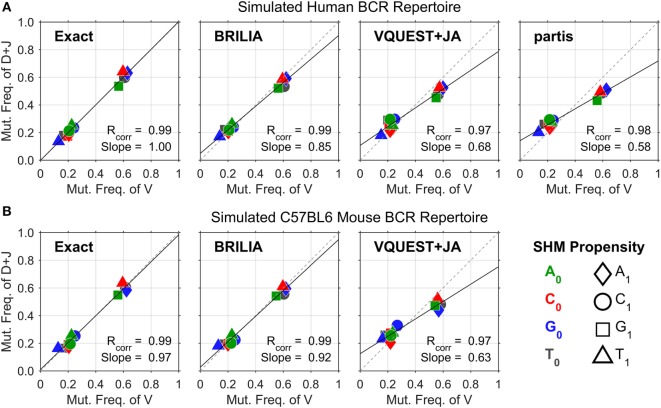
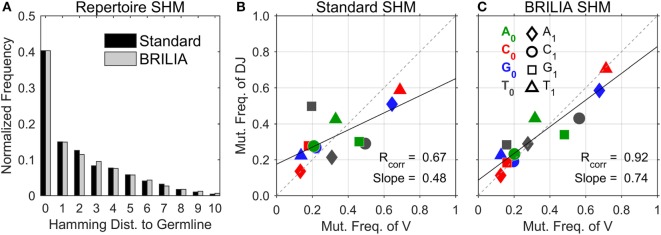
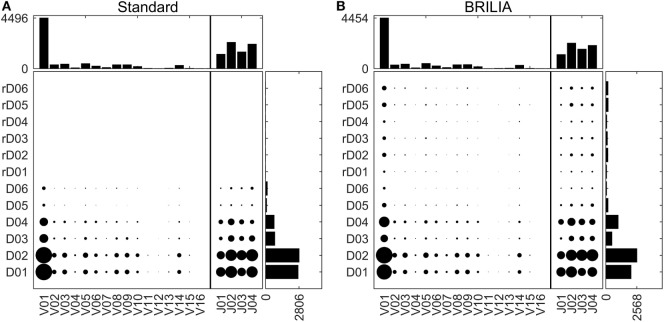
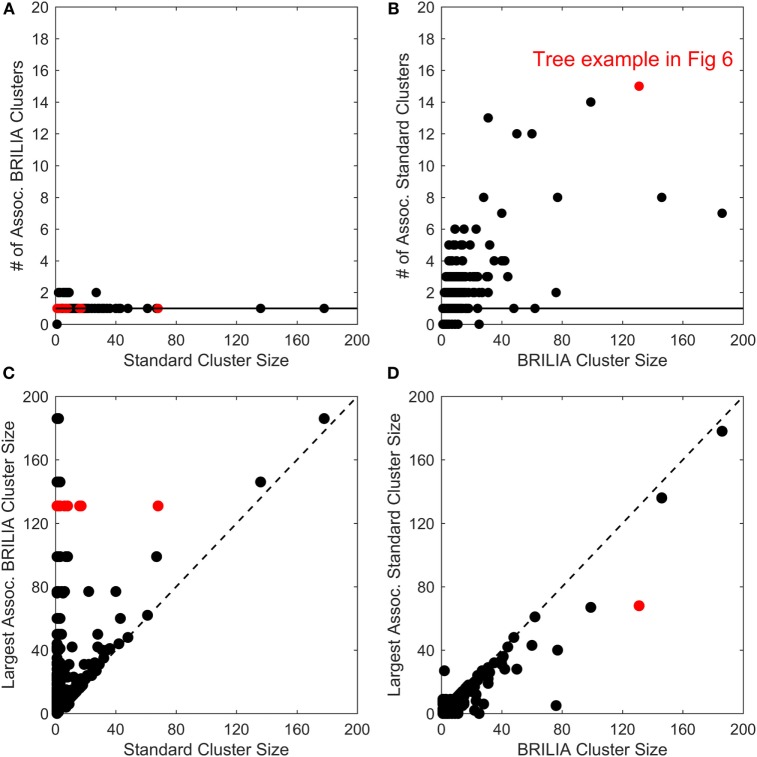
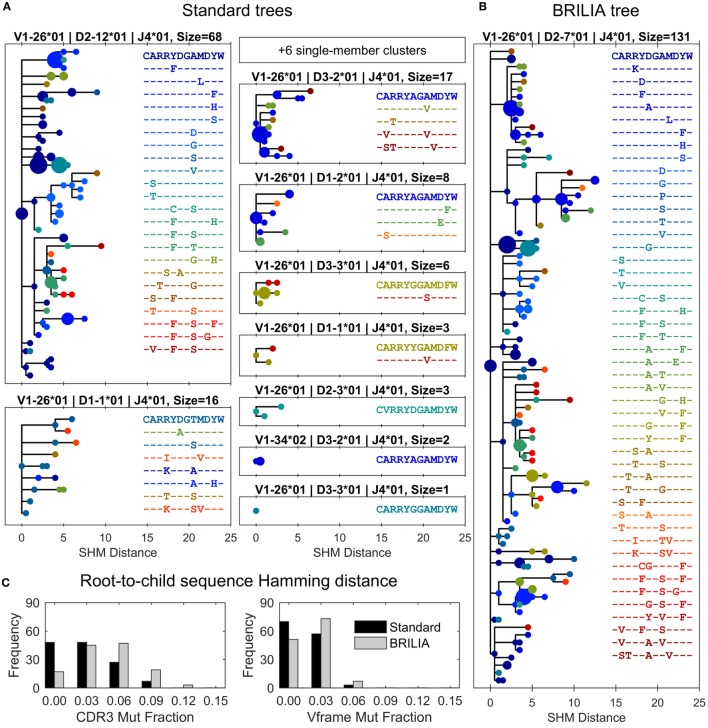
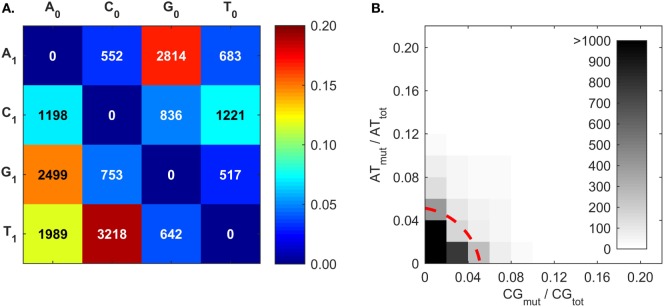
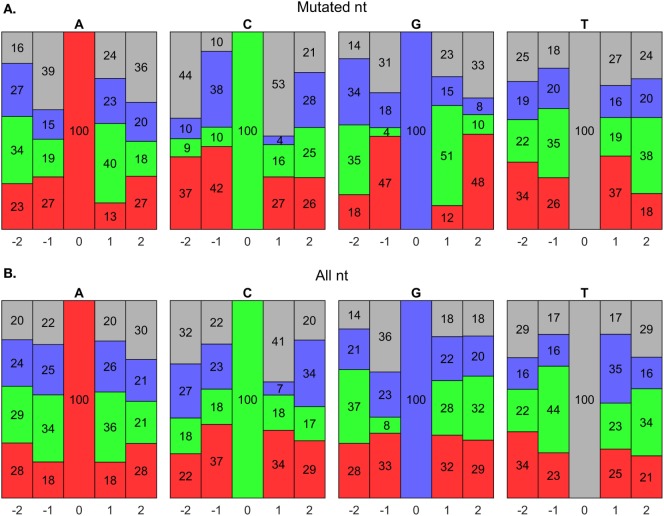
The somatic diversity of antigen-recognizing B-cell receptors (BCRs) arises from Variable (V), Diversity (D), and Joining (J) (VDJ) recombination and somatic hypermutation (SHM) during B-cell development and affinity maturation. The VDJ junction of the BCR heavy chain forms the highly variable complementarity determining region 3 (CDR3), which plays a critical role in antigen specificity and binding affinity. Tracking the selection and mutation of the CDR3 can be useful in characterizing humoral responses to infection and vaccination. Although tens to hundreds of thousands of unique BCR genes within an expressed B-cell repertoire can now be resolved with high-throughput sequencing, tracking SHMs is still challenging because existing annotation methods are often limited by poor annotation coverage, inconsistent SHM identification across the VDJ junction, or lack of B-cell lineage data. Here, we present B-cell repertoire inductive lineage and immunosequence annotator (BRILIA), an algorithm that leverages repertoire-wide sequencing data to globally improve the VDJ annotation coverage, lineage tree assembly, and SHM identification. On benchmark tests against simulated human and mouse BCR repertoires, BRILIA correctly annotated germline and clonally expanded sequences with 94 and 70% accuracy, respectively, and it has a 90% SHM-positive prediction rate in the CDR3 of heavily mutated sequences; these are substantial improvements over existing methods. We used BRILIA to process BCR sequences obtained from splenic germinal center B cells extracted from C57BL/6 mice. BRILIA returned robust B-cell lineage trees and yielded SHM patterns that are consistent across the VDJ junction and agree with known biological mechanisms of SHM. By contrast, existing BCR annotation tools, which do not account for repertoire-wide clonal relationships, systematically underestimated both the size of clonally related B-cell clusters and yielded inconsistent SHM frequencies. We demonstrate BRILIA's utility in B-cell repertoire studies related to VDJ gene usage, mechanisms for adenosine mutations, and SHM hot spot motifs. Furthermore, we show that the complete gene usage annotation and SHM identification across the entire CDR3 are essential for studying the B-cell affinity maturation process through immunosequencing methods.
抗原识别性B细胞受体(BCR)的体细胞多样性源于B细胞发育和亲和力成熟过程中的可变(V)、多样(D)和连接(J)(VDJ)重组以及体细胞超突变(SHM)。BCR重链的VDJ连接形成高度可变的互补决定区3(CDR3),其在抗原特异性和结合亲和力方面起着关键作用。追踪CDR3的选择和突变有助于表征对感染和疫苗接种的体液免疫反应。尽管现在通过高通量测序可以解析表达的B细胞库中数万至数十万独特的BCR基因,但追踪SHM仍然具有挑战性,因为现有的注释方法通常受到注释覆盖率低、VDJ连接上SHM识别不一致或缺乏B细胞谱系数据的限制。在此,我们提出了B细胞库诱导谱系和免疫序列注释器(BRILIA),这是一种利用全库测序数据来全面提高VDJ注释覆盖率、谱系树组装和SHM识别的算法。在针对模拟的人类和小鼠BCR库的基准测试中,BRILIA分别以94%和70%的准确率正确注释了种系和克隆扩增序列,并且在高度突变序列的CDR3中具有90%的SHM阳性预测率;这些都是相对于现有方法的显著改进。我们使用BRILIA处理从C57BL/6小鼠脾脏生发中心B细胞获得的BCR序列。BRILIA返回了稳健的B细胞谱系树,并产生了在VDJ连接上一致且与已知SHM生物学机制相符的SHM模式。相比之下,现有的不考虑全库克隆关系的BCR注释工具,系统性地低估了克隆相关B细胞簇的大小,并且产生了不一致的SHM频率。我们展示了BRILIA在与VDJ基因使用、腺苷突变机制和SHM热点基序相关的B细胞库研究中的效用。此外,我们表明通过免疫测序方法研究B细胞亲和力成熟过程时,整个CDR3的完整基因使用注释和SHM识别至关重要。