Department of Oncology, Zhongda Hospital, School of Medicine, Southeast University, Nanjing, China.
Pathol Oncol Res. 2022 Aug 19;28:1610504. doi: 10.3389/pore.2022.1610504. eCollection 2022.
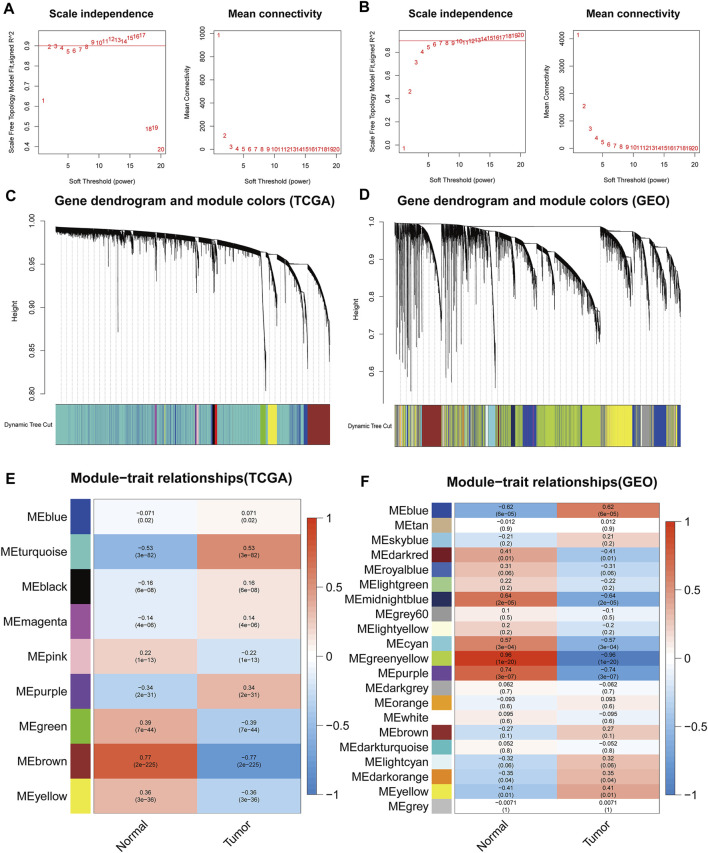
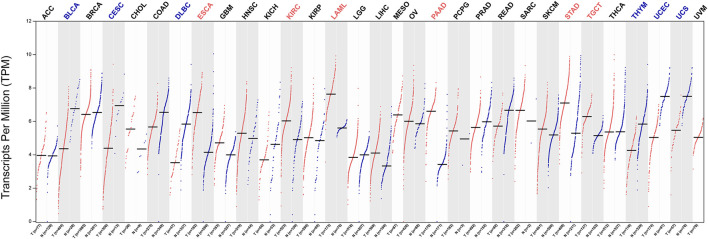
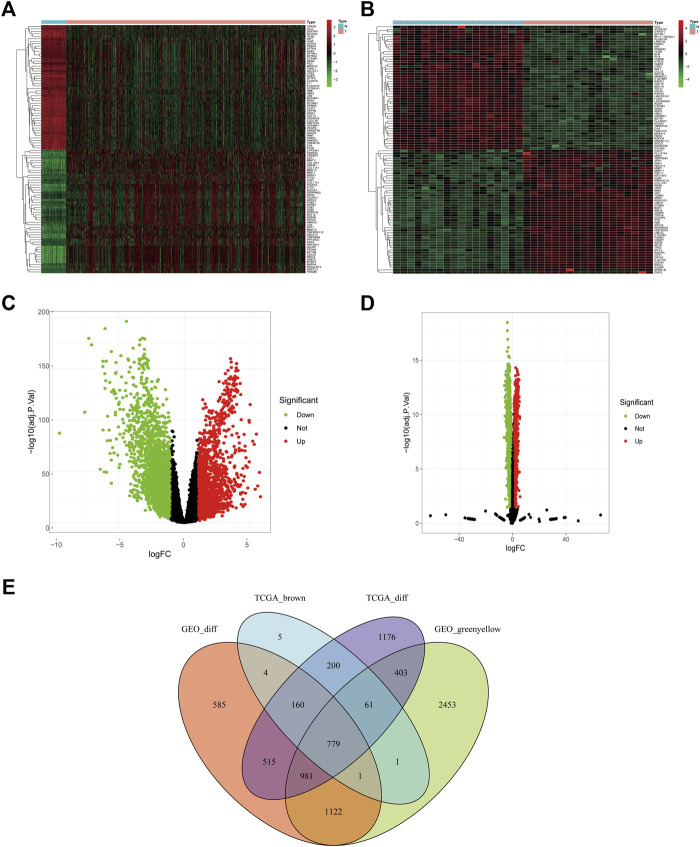
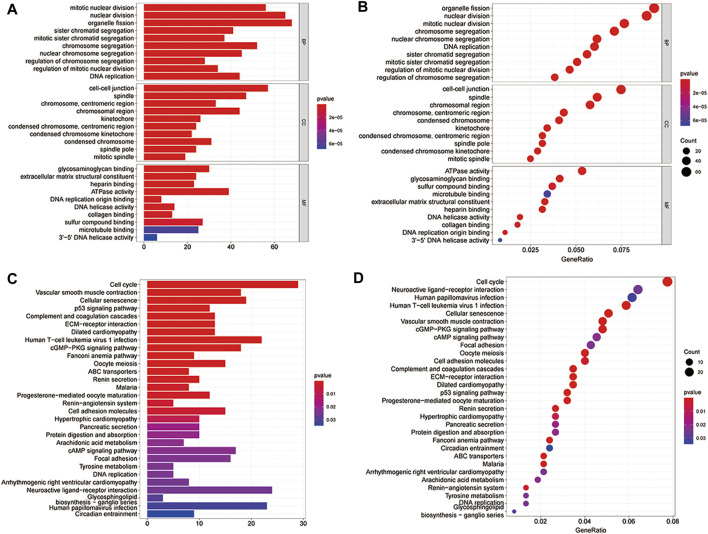
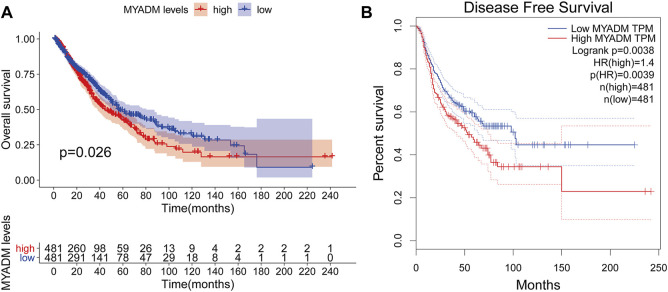
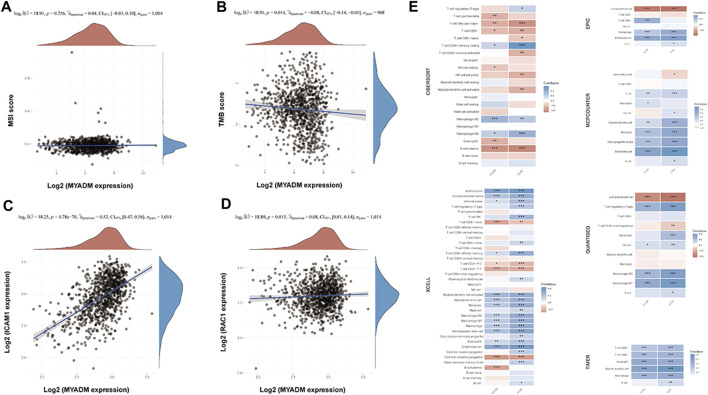
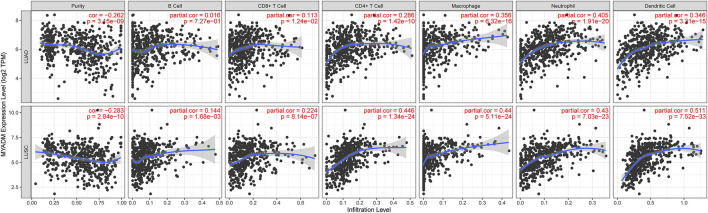
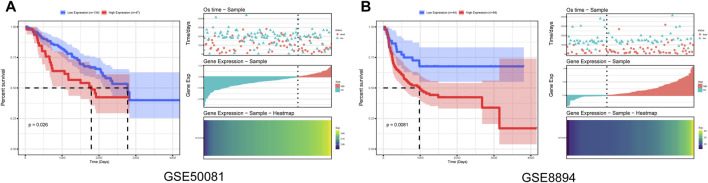
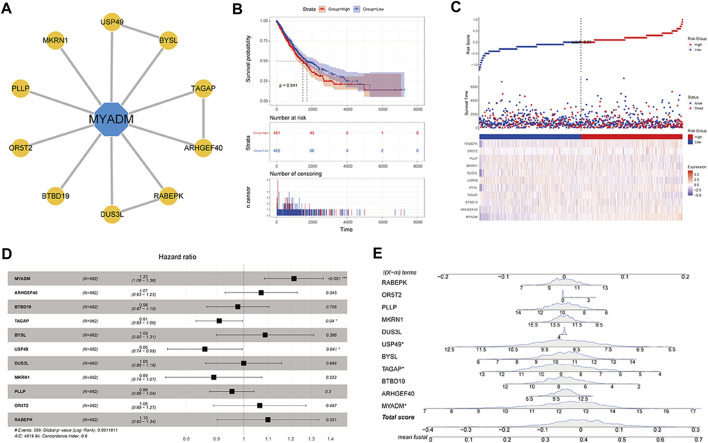
This study aimed to identify a molecular marker associated with the prognosis of non-small-cell lung cancer (NSCLC). The RNA sequencing data and clinical information of NSCLC patients were obtained from The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO). The weighted gene co-expression network analysis (WGCNA) was used to identify the co-expression gene modules and differentially expressed genes (DEGs) by comparing gene expression between NSCLC tumor tissues and normal tissues. Subsequently, the functional enrichment analysis of the DEGs was performed. Kaplan-Meier survival analysis and the GEPIA2 online tool were performed to investigate the relationship between the expression of these genes of interest and the survival of NSCLC patients, and to validate one most survival-relevent hub gene, as well as validated the hub gene using independent datasets from the GEO database. Further analysis was carried out to characterize the relationship between the hub gene and tumor immune cell infiltration, tumor mutation burden (TMB), microsatellite instability (MSI), and other known biomarkers of lung cancer. The related genes were screened by analyzing the protein-protein interaction (PPI) network and the survival model was constructed. GEPIA2 was applied in the potential analysis of pan-cancer biomarker of hub gene. 57 hub genes were found to be involved in intercellular connectivity from the 779 identified differentially co-expressed genes. Myeloid-associated differentiation marker () was strongly associated with overall survival (OS) and disease-free survival (DFS) of NSCLC patients, and high expression was associated with poor prognosis. Thus, was identified as a risk factor. Additionally, was validated as a survival risk factor in NSCLC patients in two independent datasets. Further analysis showed that was nagetively associated with TMB, and was positively correlated with macrophages, neutrophils, and dendritic cells, suggesting its role in regulating tumor immunity. The expression differed across many types of cancer and had the potential to serve as a pan-cancer marker. is an independent prognostic factor for NSCLC patients, which can predict the progression of cancer and play a role in the tumor immune cell infiltration in NSCLC.
本研究旨在鉴定与非小细胞肺癌(NSCLC)预后相关的分子标志物。从癌症基因组图谱(TCGA)和基因表达综合数据库(GEO)中获取 NSCLC 患者的 RNA 测序数据和临床信息。通过比较 NSCLC 肿瘤组织和正常组织中的基因表达,采用加权基因共表达网络分析(WGCNA)鉴定共表达基因模块和差异表达基因(DEGs)。随后,对 DEGs 进行功能富集分析。通过 Kaplan-Meier 生存分析和 GEPIA2 在线工具,研究这些感兴趣基因的表达与 NSCLC 患者生存之间的关系,并验证一个最具生存相关性的枢纽基因,同时使用 GEO 数据库中的独立数据集验证该枢纽基因。进一步分析以表征枢纽基因与肿瘤免疫细胞浸润、肿瘤突变负荷(TMB)、微卫星不稳定性(MSI)和其他已知肺癌生物标志物之间的关系。通过分析蛋白质-蛋白质相互作用(PPI)网络筛选相关基因,并构建生存模型。应用 GEPIA2 对枢纽基因的泛癌生物标志物进行潜在分析。从鉴定的 779 个差异共表达基因中筛选出 57 个与细胞间连接性相关的枢纽基因。髓系相关分化标志物()与 NSCLC 患者的总生存(OS)和无病生存(DFS)密切相关,高表达与预后不良相关,因此被鉴定为危险因素。此外,在两个独立的数据集验证了 是 NSCLC 患者的生存危险因素。进一步分析表明,与 TMB 呈负相关,与巨噬细胞、中性粒细胞和树突状细胞呈正相关,提示其在调节肿瘤免疫中的作用。在多种类型的癌症中,表达存在差异,具有作为泛癌标志物的潜力。 是 NSCLC 患者的独立预后因素,可预测癌症的进展,并在 NSCLC 肿瘤免疫细胞浸润中发挥作用。