Department of Human Genetics OE 6300, Hannover Medical School, Carl-Neuberg-Str. 1, 30625, Hannover, Germany.
Department of Pediatric Kidney, Liver and Metabolic Diseases, Hannover Medical School, Hannover, Germany.
Hum Genet. 2023 Jan;142(1):73-88. doi: 10.1007/s00439-022-02481-6. Epub 2022 Sep 6.
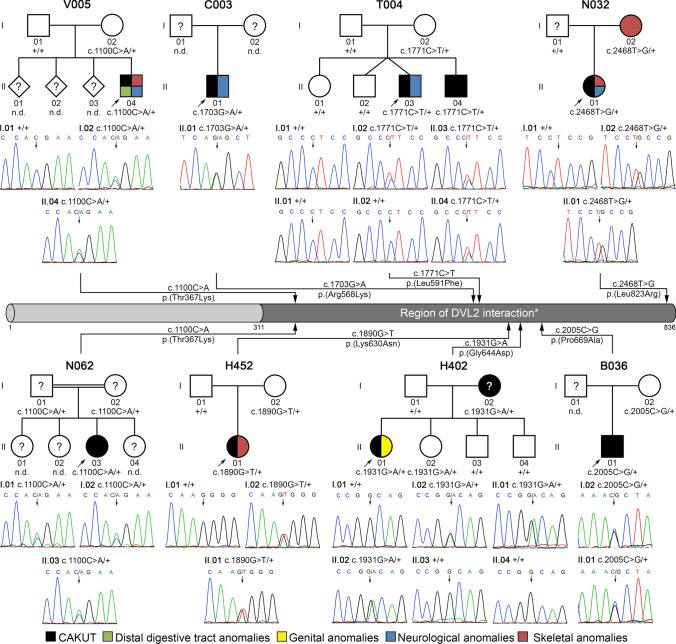
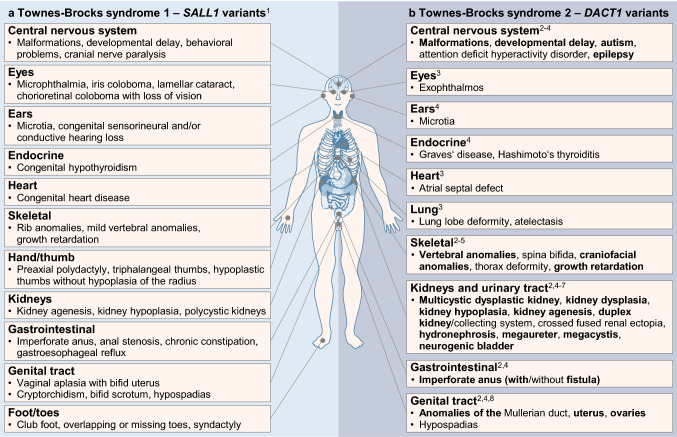
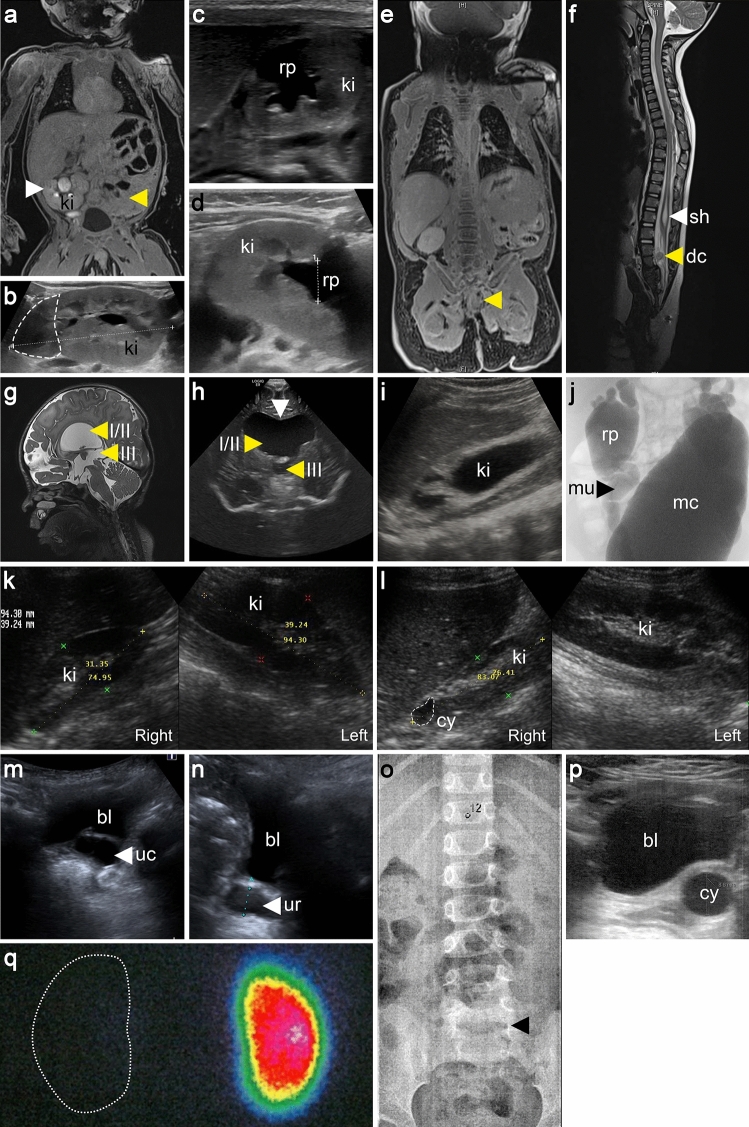
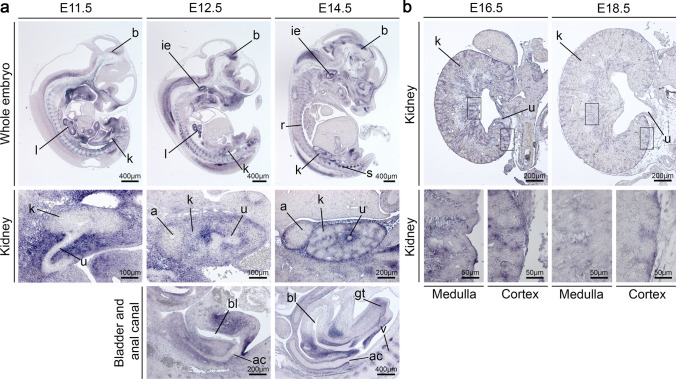
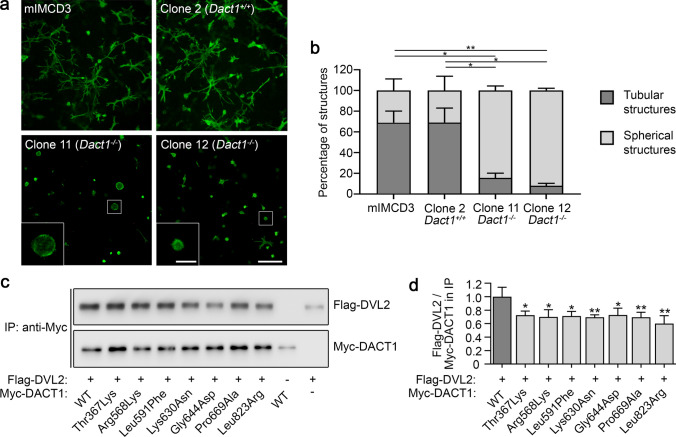
Most patients with congenital anomalies of the kidney and urinary tract (CAKUT) remain genetically unexplained. In search of novel genes associated with CAKUT in humans, we applied whole-exome sequencing in a patient with kidney, anorectal, spinal, and brain anomalies, and identified a rare heterozygous missense variant in the DACT1 (dishevelled binding antagonist of beta catenin 1) gene encoding a cytoplasmic WNT signaling mediator. Our patient's features overlapped Townes-Brocks syndrome 2 (TBS2) previously described in a family carrying a DACT1 nonsense variant as well as those of Dact1-deficient mice. Therefore, we assessed the role of DACT1 in CAKUT pathogenesis. Taken together, very rare (minor allele frequency ≤ 0.0005) non-silent DACT1 variants were detected in eight of 209 (3.8%) CAKUT families, significantly more frequently than in controls (1.7%). All seven different DACT1 missense variants, predominantly likely pathogenic and exclusively maternally inherited, were located in the interaction region with DVL2 (dishevelled segment polarity protein 2), and biochemical characterization revealed reduced binding of mutant DACT1 to DVL2. Patients carrying DACT1 variants presented with kidney agenesis, duplex or (multi)cystic (hypo)dysplastic kidneys with hydronephrosis and TBS2 features. During murine development, Dact1 was expressed in organs affected by anomalies in patients with DACT1 variants, including the kidney, anal canal, vertebrae, and brain. In a branching morphogenesis assay, tubule formation was impaired in CRISPR/Cas9-induced Dact1 murine inner medullary collecting duct cells. In summary, we provide evidence that heterozygous hypomorphic DACT1 variants cause CAKUT and other features of TBS2, including anomalies of the skeleton, brain, distal digestive and genital tract.
大多数先天性肾和尿路畸形(CAKUT)患者的遗传原因仍不清楚。为了寻找与人类 CAKUT 相关的新基因,我们对一名患有肾、肛门直肠、脊柱和脑异常的患者进行了全外显子组测序,发现了一个罕见的杂合错义变异,该变异位于编码细胞质 WNT 信号转导介质的 DACT1(dishevelled binding antagonist of beta catenin 1)基因中。我们患者的特征与 Townes-Brocks 综合征 2(TBS2)重叠,该综合征以前在一个携带 DACT1 无义变异的家族中描述过,也与 Dact1 缺陷小鼠的特征重叠。因此,我们评估了 DACT1 在 CAKUT 发病机制中的作用。总的来说,在 209 个 CAKUT 家族中的 8 个(3.8%)中检测到非常罕见的(次要等位基因频率≤0.0005)非沉默 DACT1 变异,明显高于对照组(1.7%)。所有 7 种不同的 DACT1 错义变异均位于与 DVL2(dishevelled segment polarity protein 2)相互作用的区域,主要为可能致病性且仅为母系遗传,生化特征显示突变型 DACT1 与 DVL2 的结合减少。携带 DACT1 变异的患者表现为肾发育不全、双肾盂或(多)囊性(低)发育不良肾伴肾积水和 TBS2 特征。在小鼠发育过程中,Dact1 在患者携带 DACT1 变异的受影响器官中表达,包括肾脏、肛门直肠、脊椎和大脑。在分支形态发生测定中,CRISPR/Cas9 诱导的 Dact1 小鼠内髓集合管细胞的管形成受损。总之,我们提供的证据表明,杂合功能降低的 DACT1 变异导致 CAKUT 和 TBS2 的其他特征,包括骨骼、大脑、远端消化和生殖道的异常。