CICS-UBI, Health Sciences Research Centre, University of Beira Interior, 6200-506 Covilha, Portugal.
Serviço de Endocrinologia, Diabetes e Metabolismo, Centro Hospitalar Universitário de Coimbra, 3000-075 Coimbra, Portugal.
Int J Mol Sci. 2022 Sep 2;23(17):10026. doi: 10.3390/ijms231710026.
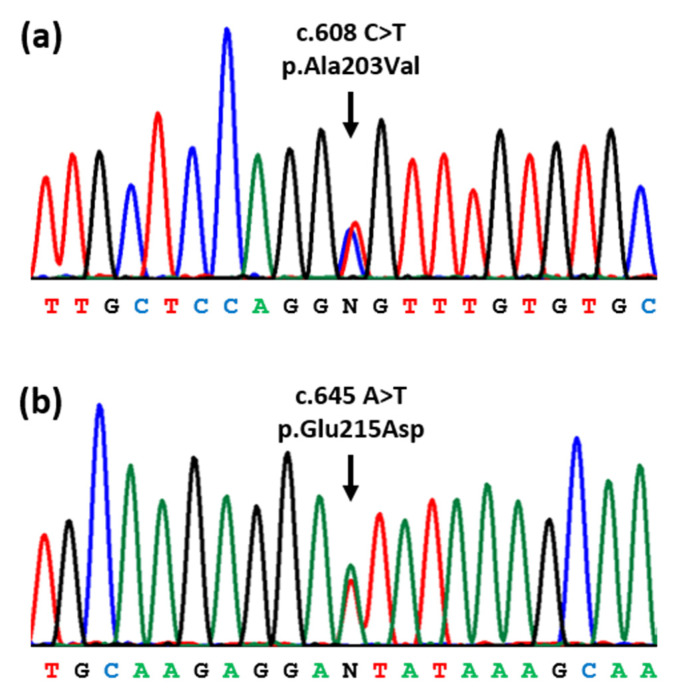
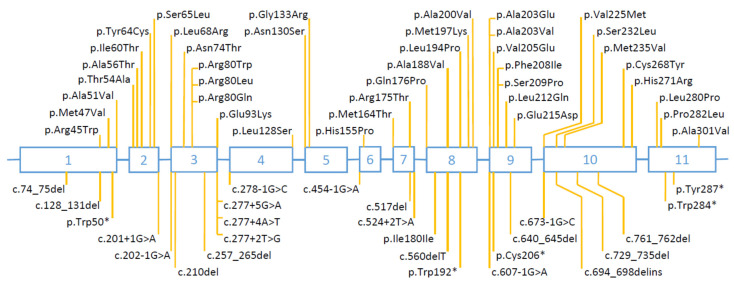
The 17-beta-hydroxysteroid dehydrogenase type 3 (17-β-HSD3) enzyme converts androstenedione to testosterone and is encoded by the HSD17B3 gene. Homozygous or compound heterozygous HSD17B3 mutations block the synthesis of testosterone in the fetal testis, resulting in a Disorder of Sex Development (DSD). We describe a child raised as a female in whom the discovery of testes in the inguinal canals led to a genetic study by whole exome sequencing (WES) and to the identification of a compound heterozygous mutation of the HSD17B3 gene (c.608C>T, p.Ala203Val, and c.645A>T, p.Glu215Asp). Furthermore, we review all HSD17B3 mutations published so far in cases of 17-β-HSD3 deficiency. A total of 70 different HSD17B3 mutations have so far been reported in 239 patients from 187 families. A total of 118 families had homozygous mutations, 63 had compound heterozygous mutations and six had undetermined genotypes. Mutations occurred in all 11 exons and were missense (55%), splice-site (29%), small deletions and insertions (7%), nonsense (5%), and multiple exon deletions and duplications (2%). Several mutations were recurrent and missense mutations at codon 80 and the splice-site mutation c.277+4A>T each represented 17% of all mutated alleles. These findings may be useful to those involved in the clinical management and genetic diagnosis of this disorder.
17-β-羟类固醇脱氢酶 3 型(17-β-HSD3)酶将雄烯二酮转化为睾酮,由 HSD17B3 基因编码。HSD17B3 基因的纯合子或复合杂合突变会阻止胎儿睾丸中睾酮的合成,导致性发育障碍(DSD)。我们描述了一名被作为女性抚养的儿童,由于在腹股沟管中发现了睾丸,进行了全外显子组测序(WES)的遗传研究,并确定了 HSD17B3 基因的复合杂合突变(c.608C>T,p.Ala203Val 和 c.645A>T,p.Glu215Asp)。此外,我们回顾了迄今为止所有发表的 HSD17B3 突变病例,这些病例都与 17-β-HSD3 缺乏有关。迄今为止,在 187 个家族的 239 名患者中,共报道了 70 种不同的 HSD17B3 突变。共有 118 个家系为纯合突变,63 个家系为复合杂合突变,6 个家系为未确定基因型。突变发生在所有 11 个外显子中,为错义突变(55%)、剪接位点突变(29%)、小的缺失和插入(7%)、无义突变(5%)和多个外显子缺失和重复(2%)。一些突变是复发性的,密码子 80 处的错义突变和剪接位点突变 c.277+4A>T 分别占所有突变等位基因的 17%。这些发现可能对参与该疾病的临床管理和遗传诊断的人员有用。