National Institute of Chemistry, Hajdrihova 19, SI-1001, Ljubljana, Slovenia.
Jozef Stefan International Postgraduate School, Jamova cesta 39, 1000, Ljubljana, Slovenia.
Genome Biol. 2022 Sep 9;23(1):191. doi: 10.1186/s13059-022-02755-2.
Crosslinking and immunoprecipitation (CLIP) is a method used to identify in vivo RNA-protein binding sites on a transcriptome-wide scale. With the increasing amounts of available data for RNA-binding proteins (RBPs), it is important to understand to what degree the enriched motifs specify the RNA-binding profiles of RBPs in cells.
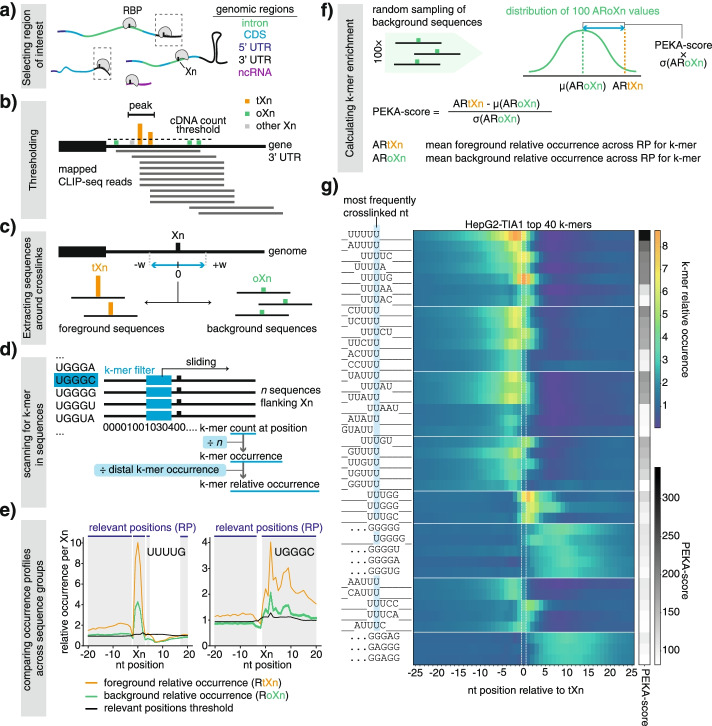
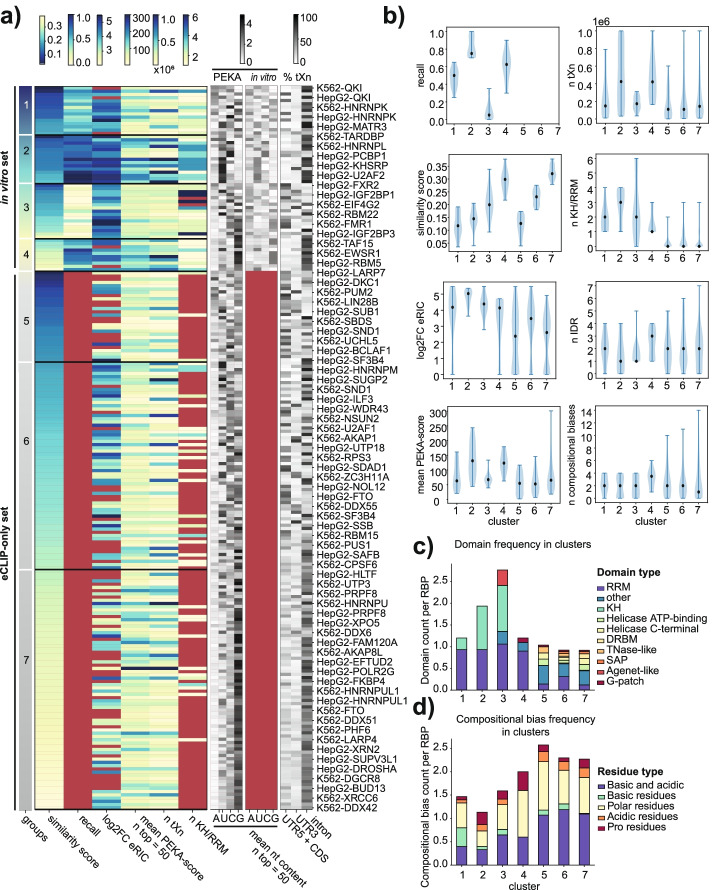
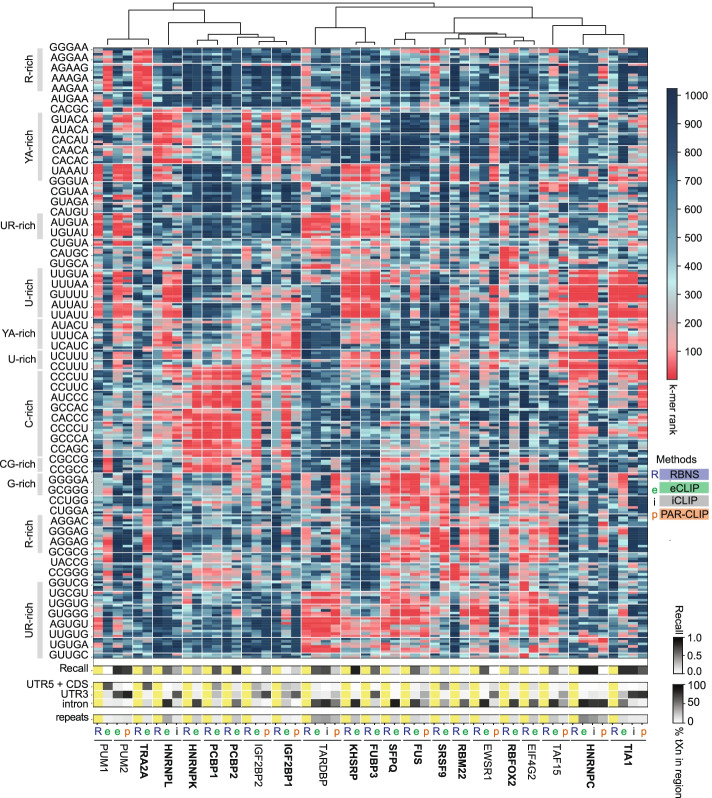
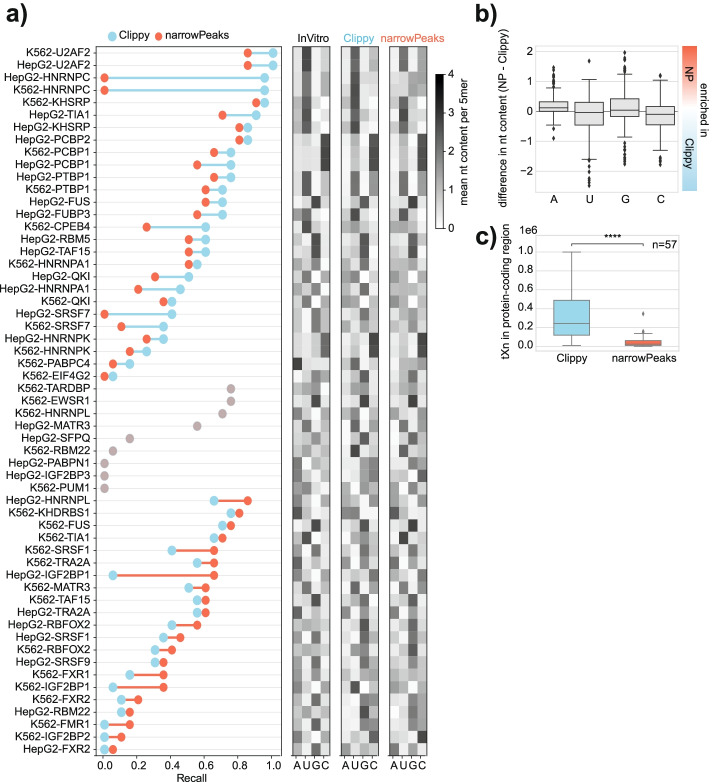
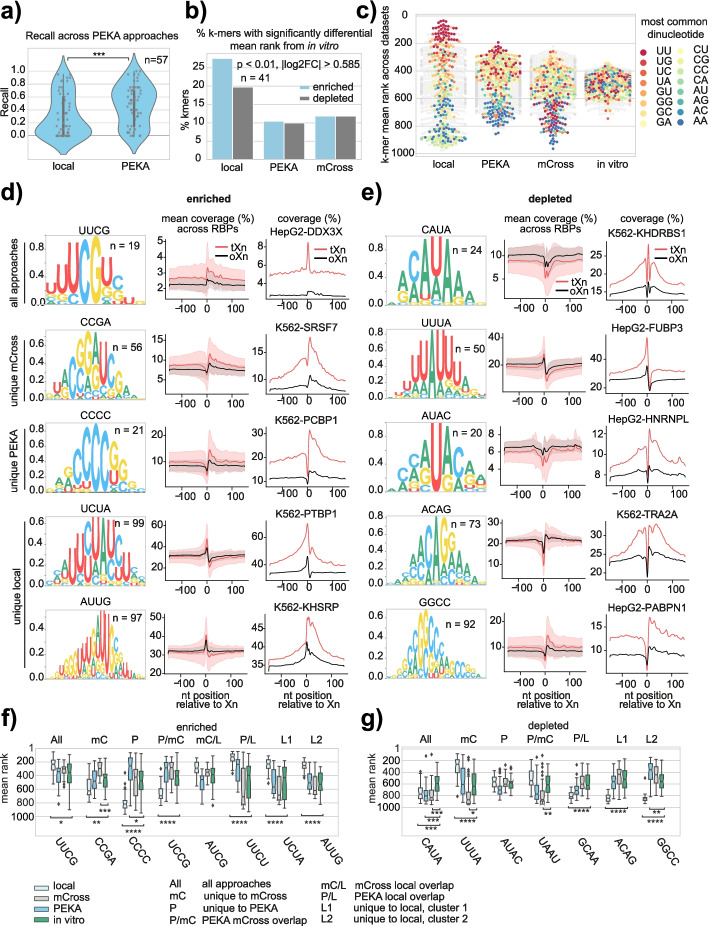
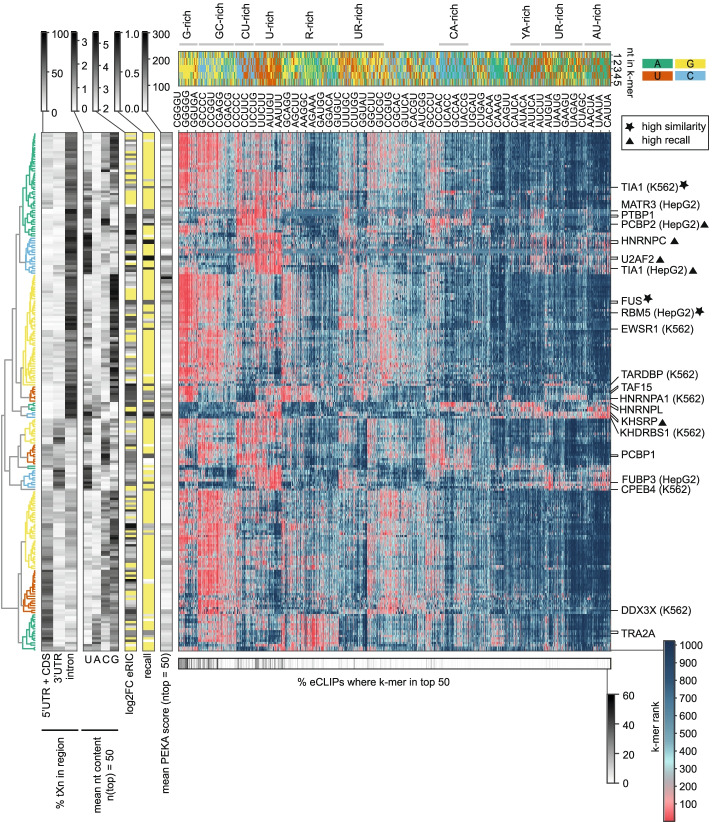
We develop positionally enriched k-mer analysis (PEKA), a computational tool for efficient analysis of enriched motifs from individual CLIP datasets, which minimizes the impact of technical and regional genomic biases by internal data normalization. We cross-validate PEKA with mCross and show that the use of input control for background correction is not required to yield high specificity of enriched motifs. We identify motif classes with common enrichment patterns across eCLIP datasets and across RNA regions, while also observing variations in the specificity and the extent of motif enrichment across eCLIP datasets, between variant CLIP protocols, and between CLIP and in vitro binding data. Thereby, we gain insights into the contributions of technical and regional genomic biases to the enriched motifs, and find how motif enrichment features relate to the domain composition and low-complexity regions of the studied proteins.
Our study provides insights into the overall contributions of regional binding preferences, protein domains, and low-complexity regions to the specificity of protein-RNA interactions, and shows the value of cross-motif and cross-RBP comparison for data interpretation. Our results are presented for exploratory analysis via an online platform in an RBP-centric and motif-centric manner ( https://imaps.goodwright.com/apps/peka/ ).
交联和免疫沉淀(CLIP)是一种用于在转录组范围内鉴定体内 RNA-蛋白质结合位点的方法。随着 RNA 结合蛋白(RBP)可用数据量的增加,了解富集的基序在多大程度上指定 RBP 在细胞中的 RNA 结合谱变得非常重要。
我们开发了位置富集 k-mer 分析(PEKA),这是一种用于有效分析单个 CLIP 数据集富集基序的计算工具,通过内部数据归一化最小化了技术和区域基因组偏倚的影响。我们使用 mCross 对 PEKA 进行了交叉验证,结果表明,为了获得富集基序的高特异性,不需要使用输入对照进行背景校正。我们确定了在 eCLIP 数据集和 RNA 区域之间具有共同富集模式的基序类别,同时也观察到在 eCLIP 数据集之间、不同 CLIP 方案之间以及 CLIP 和体外结合数据之间,富集基序的特异性和程度存在差异。由此,我们深入了解了技术和区域基因组偏倚对富集基序的贡献,并发现了基序富集特征与研究蛋白质的结构域组成和低复杂度区域之间的关系。
我们的研究提供了对区域结合偏好、蛋白质结构域和低复杂度区域对蛋白质-RNA 相互作用特异性的总体贡献的深入了解,并展示了跨基序和跨 RBP 比较在数据解释中的价值。我们的结果以 RBP 为中心和基序为中心的方式通过在线平台进行了探索性分析(https://imaps.goodwright.com/apps/peka/)。