Liu Jiawei, Zhang Ling, Gao Jian, Zhang Baochen, Liu Xiaoli, Yang Ninghui, Liu Xiaotong, Liu Xifu, Cheng Yu
Center for Drug Innovation and Discovery, College of Life Science, Hebei Normal University, Shijiazhuang, China.
School of Chemical Technology, Shijiazhuang University, Shijiazhuang, China.
Front Pharmacol. 2022 Aug 25;13:961154. doi: 10.3389/fphar.2022.961154. eCollection 2022.
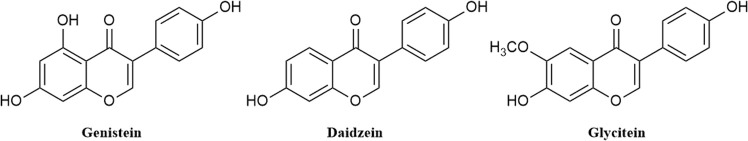
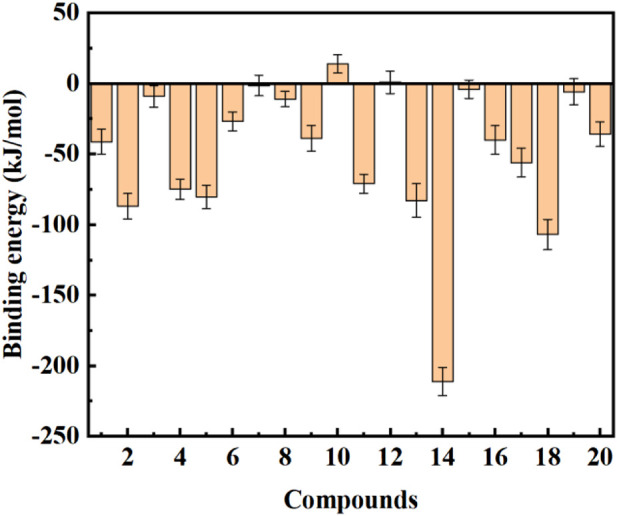
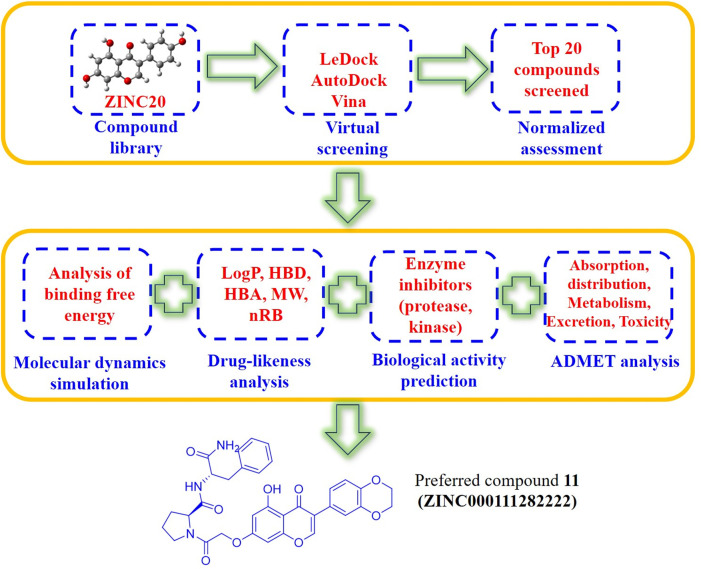
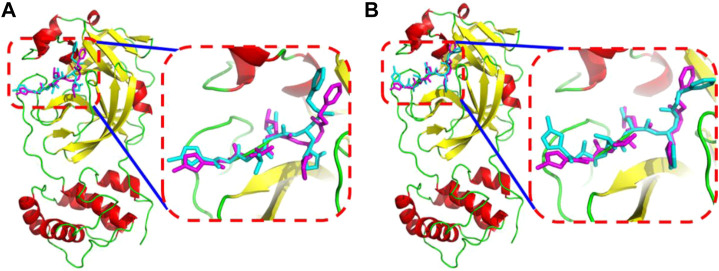
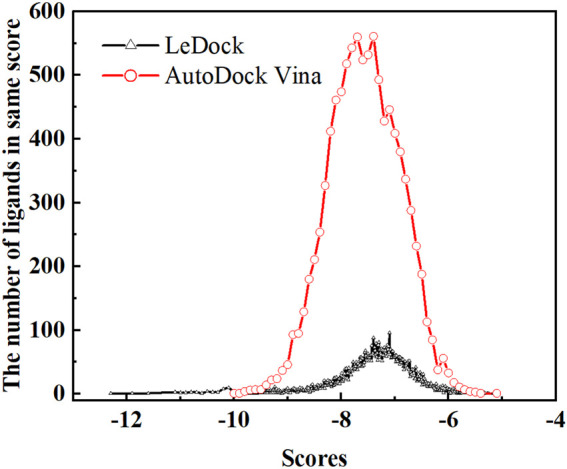
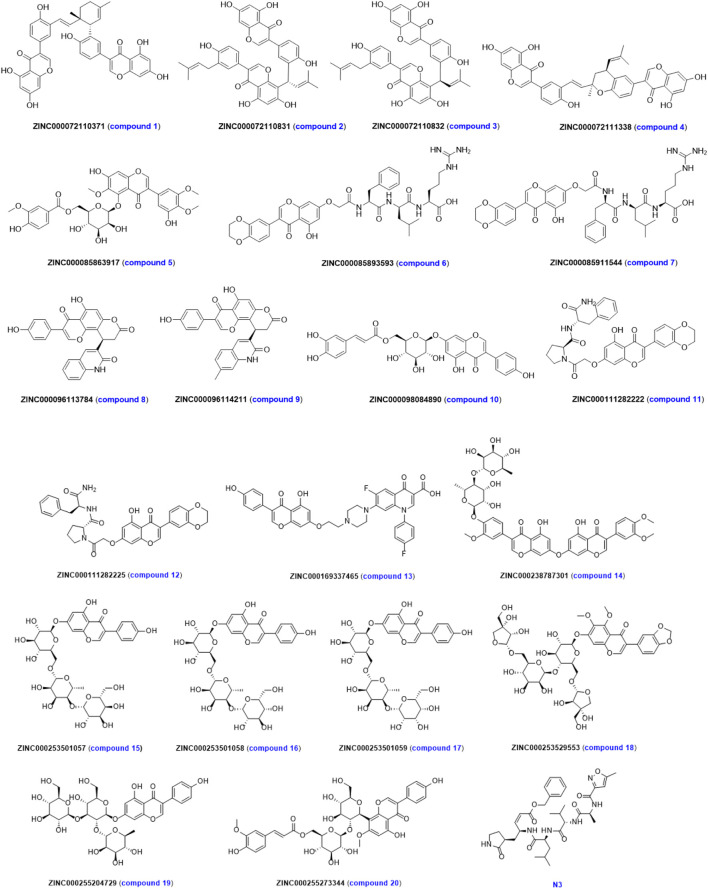
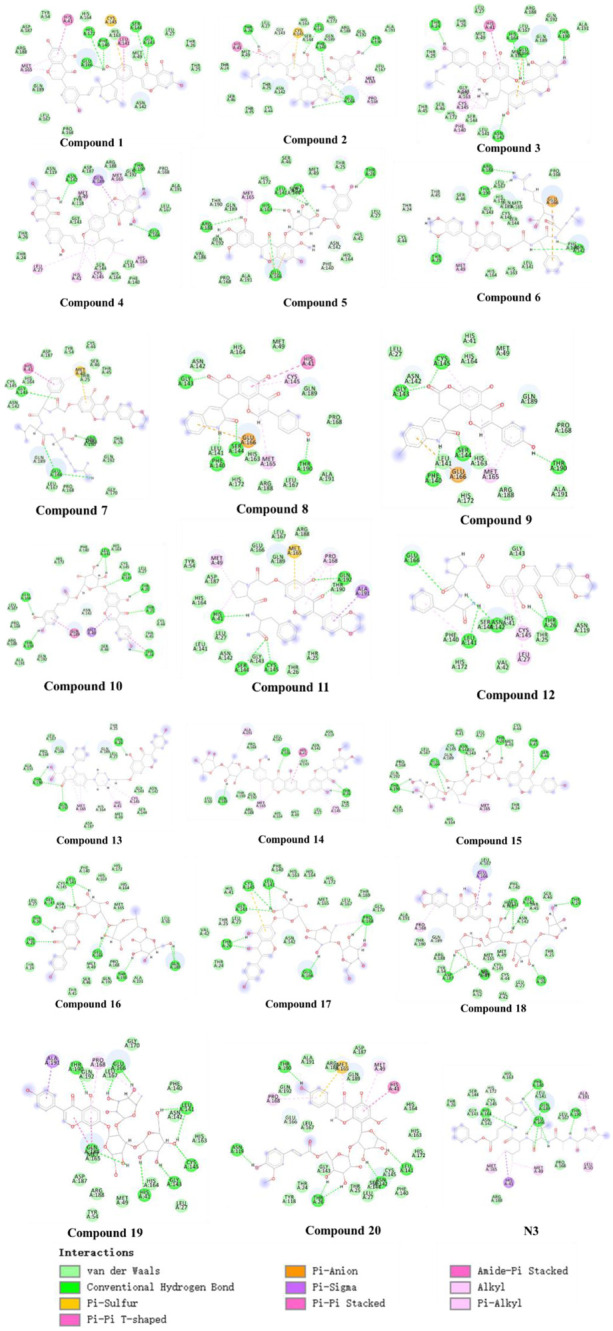
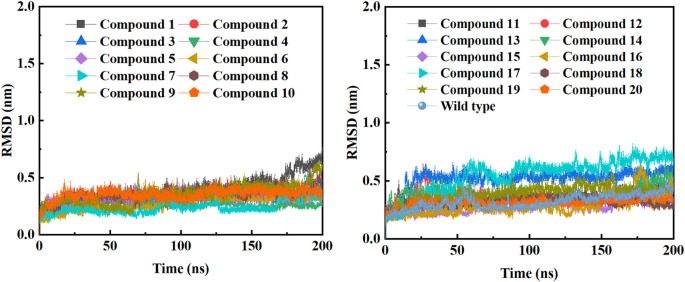
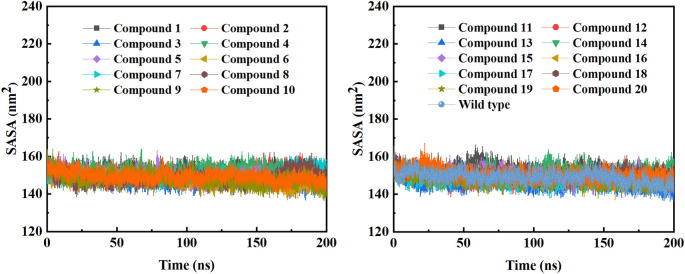
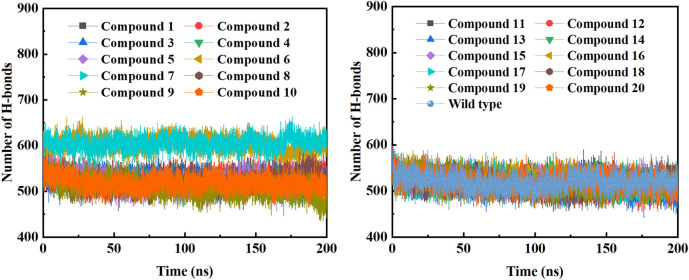
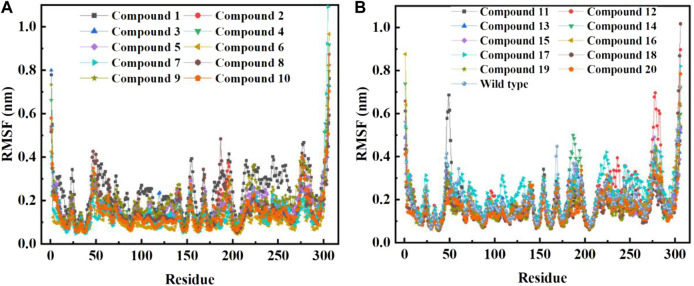
Due to the constant mutation of virus and the lack of specific therapeutic drugs, the coronavirus disease 2019 (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) still poses a huge threat to the health of people, especially those with underlying diseases. Therefore, drug discovery against the SARS-CoV-2 remains of great significance. With the main protease of virus as the inhibitor target, 9,614 genistein derivatives were virtually screened by LeDock and AutoDock Vina, and the top 20 compounds with highest normalized scores were obtained. Molecular dynamics simulations were carried out for studying interactions between these 20 compounds and the target protein. The drug-like properties, activity, and ADMET of these compounds were also evaluated by DruLiTo software or online server. Twenty compounds, including compound , were screened by normalized molecular docking, which could bind to the target through multiple non-bonding interactions. Molecular dynamics simulation results showed that compounds , , , , , , , and had the best binding force with the target protein of SARS-CoV-2, and the absolute values of binding free energies all exceeded 50 kJ/mol. The drug-likeness properties indicated that a variety of compounds including compound were worthy of further study. The results of bioactivity score prediction found that compounds and had high inhibitory activities against protease, which indicated that these two compounds had the potential to be further developed as COVID-19 inhibitors. Finally, compound showed excellent predictive ADMET properties including high absorption and low toxicity. These in silico work results show that the preferred compound (), which exhibited strong binding to SARS-CoV-2 main protease, acceptable drug-like properties, protease inhibitory activity and ADMET properties, has great promise for further research as a potential therapeutic agent against COVID-19.
由于病毒的不断变异以及缺乏特效治疗药物,由严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的2019冠状病毒病(COVID-19)大流行仍然对人们的健康构成巨大威胁,尤其是对那些患有基础疾病的人。因此,针对SARS-CoV-2的药物研发仍然具有重要意义。以病毒的主要蛋白酶为抑制剂靶点,通过LeDock和AutoDock Vina对9614种染料木黄酮衍生物进行虚拟筛选,得到归一化分数最高的前20种化合物。对这20种化合物与靶蛋白之间的相互作用进行了分子动力学模拟。还通过DruLiTo软件或在线服务器评估了这些化合物的类药性质、活性和药物代谢动力学/药物毒性学性质。通过归一化分子对接筛选出包括化合物 在内的20种化合物,它们可以通过多种非键相互作用与靶点结合。分子动力学模拟结果表明,化合物 、 、 、 、 、 、 和 与SARS-CoV-2靶蛋白的结合力最佳,结合自由能的绝对值均超过50 kJ/mol。类药性质表明包括化合物 在内的多种化合物值得进一步研究。生物活性评分预测结果发现,化合物 和 对蛋白酶具有高抑制活性,这表明这两种化合物有潜力进一步开发为COVID-19抑制剂。最后,化合物 表现出优异的预测药物代谢动力学/药物毒性学性质,包括高吸收和低毒性。这些计算机模拟工作结果表明,优选的化合物 ()与SARS-CoV-2主要蛋白酶具有强结合力、可接受的类药性质、蛋白酶抑制活性和药物代谢动力学/药物毒性学性质,作为一种潜在的抗COVID-19治疗剂具有很大的进一步研究前景。