Department of Pharmacological Sciences, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY, 10029, USA.
Department of Medicine, Korea University College of Medicine, Seoul, Republic of Korea.
BMC Bioinformatics. 2022 Sep 13;23(1):374. doi: 10.1186/s12859-022-04895-5.
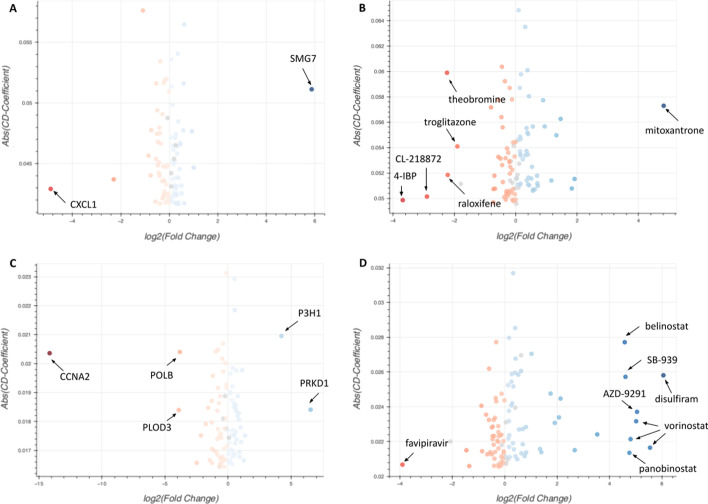
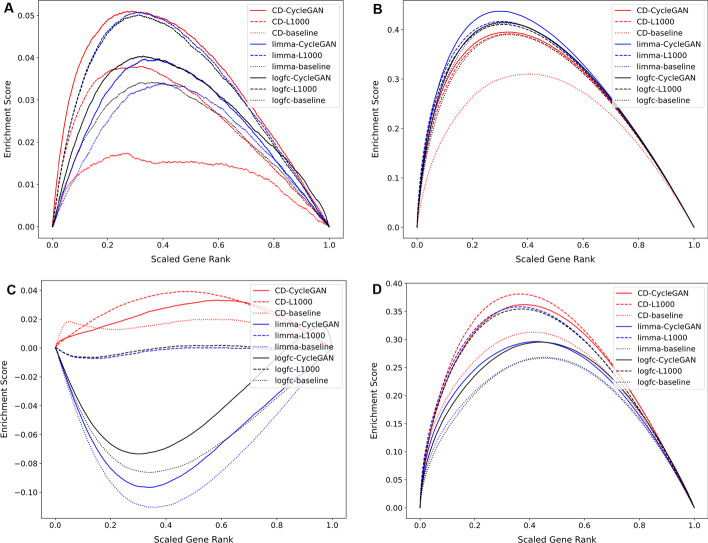
The L1000 technology, a cost-effective high-throughput transcriptomics technology, has been applied to profile a collection of human cell lines for their gene expression response to > 30,000 chemical and genetic perturbations. In total, there are currently over 3 million available L1000 profiles. Such a dataset is invaluable for the discovery of drug and target candidates and for inferring mechanisms of action for small molecules. The L1000 assay only measures the mRNA expression of 978 landmark genes while 11,350 additional genes are computationally reliably inferred. The lack of full genome coverage limits knowledge discovery for half of the human protein coding genes, and the potential for integration with other transcriptomics profiling data. Here we present a Deep Learning two-step model that transforms L1000 profiles to RNA-seq-like profiles. The input to the model are the measured 978 landmark genes while the output is a vector of 23,614 RNA-seq-like gene expression profiles. The model first transforms the landmark genes into RNA-seq-like 978 gene profiles using a modified CycleGAN model applied to unpaired data. The transformed 978 RNA-seq-like landmark genes are then extrapolated into the full genome space with a fully connected neural network model. The two-step model achieves 0.914 Pearson's correlation coefficients and 1.167 root mean square errors when tested on a published paired L1000/RNA-seq dataset produced by the LINCS and GTEx programs. The processed RNA-seq-like profiles are made available for download, signature search, and gene centric reverse search with unique case studies.
L1000 技术是一种具有成本效益的高通量转录组学技术,已被用于分析一系列人类细胞系对超过 30000 种化学和遗传扰动的基因表达反应。目前,总共有超过 300 万个可用的 L1000 图谱。对于发现药物和靶标候选物以及推断小分子的作用机制,这样的数据集是非常宝贵的。L1000 测定法仅测量 978 个标志性基因的 mRNA 表达,而另外 11350 个基因则通过计算可靠地推断。全基因组覆盖的缺乏限制了人类蛋白编码基因的一半的知识发现,以及与其他转录组学分析数据整合的潜力。在这里,我们提出了一个深度学习两步模型,将 L1000 图谱转换为 RNA-seq 样图谱。该模型的输入是测量的 978 个标志性基因,而输出是一个 23614 个 RNA-seq 样基因表达图谱的向量。该模型首先使用应用于非配对数据的修改后的 CycleGAN 模型将标志性基因转换为 RNA-seq 样的 978 个基因图谱。然后,通过全连接神经网络模型将转换后的 978 个 RNA-seq 样标志性基因外推到全基因组空间。在经过 LINCS 和 GTEx 项目生成的已发表的配对 L1000/RNA-seq 数据集上进行测试时,两步模型达到了 0.914 的皮尔逊相关系数和 1.167 的均方根误差。处理后的 RNA-seq 样图谱可用于下载、签名搜索和以独特的案例研究为中心的基因反向搜索。