Alanazi Wael A, Alhamami Hussain N, Alharbi Metab, Alhazzani Khalid, Alanazi Abdulrahman S, Alsanea Sary, Ali Nemat, Alasmari Abdullah F, Alanazi Ahmed Z, Alotaibi Moureq R, Alswayyed Mohammed
Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia.
Department of Pathology, College of Medicine, King Saud University, Riyadh 11451, Saudi Arabia.
Saudi Pharm J. 2022 Aug;30(8):1159-1169. doi: 10.1016/j.jsps.2022.06.020. Epub 2022 Jun 22.
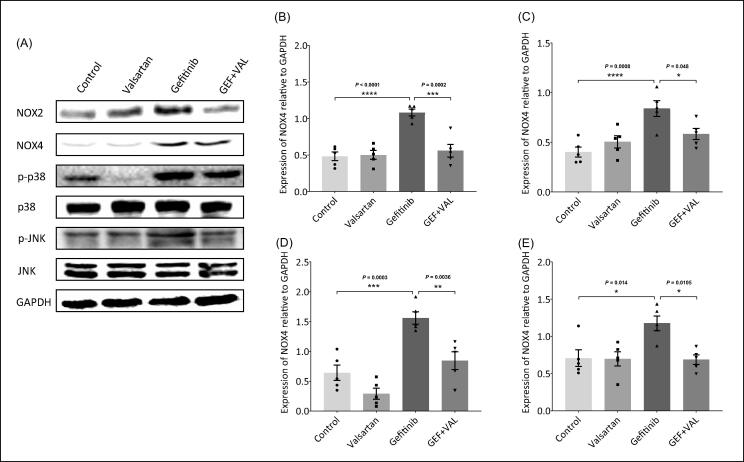
Gefitinib is a tyrosine kinase inhibitor (TKI) of the epidermal growth factor receptor (EGFR), used for the treatment of advanced or metastatic non-small cell lung cancer. Recently, studies proved that Gefitinib-induced cardiotoxicity through induction of oxidative stress leads to cardiac hypertrophy. The current study was conducted to understand the mechanisms underlying gefitinib-induced cardiac hypertrophy through studying the roles of angiotensin II (AngII), oxidative stress, and mitogen-activated protein kinase (MAPK) pathway. Male Wistar albino rats were treated with valsartan, gefitinib, or both for four weeks. Blood samples were collected for AngII and cardiac markers measurement, and hearts were harvested for histological study and biochemical analysis. Gefitinib caused histological changes in the cardiac tissues and increased levels of cardiac hypertrophy markers, AngII and its receptors. Blocking of AngII type 1 receptor (AT1R) via valsartan protected hearts and normalized cardiac markers, AngII levels, and the expression of its receptors during gefitinib treatment. valsartan attenuated gefitinib-induced NADPH oxidase and oxidative stress leading to down-regulation of JNK/p38-MAPK pathway. Collectively, AT1R blockade adjusted AngII-induced NADPH oxidase and JNK/p38-MAPK leading to attenuation of gefitinib-induced cardiac hypertrophy. This study found a pivotal role of AngII/AT1R signaling in gefitinib-induced cardiac hypertrophy, which may provide novel approaches in the management of EGFRIs-induced cardiotoxicity.
吉非替尼是一种表皮生长因子受体(EGFR)的酪氨酸激酶抑制剂(TKI),用于治疗晚期或转移性非小细胞肺癌。最近,研究证明吉非替尼通过诱导氧化应激导致心脏毒性,进而引发心脏肥大。本研究旨在通过研究血管紧张素II(AngII)、氧化应激和丝裂原活化蛋白激酶(MAPK)途径的作用,来了解吉非替尼诱导心脏肥大的潜在机制。将雄性Wistar白化大鼠用缬沙坦、吉非替尼或两者联合处理四周。采集血样用于检测AngII和心脏标志物,摘取心脏进行组织学研究和生化分析。吉非替尼导致心脏组织发生组织学变化,并增加了心脏肥大标志物、AngII及其受体的水平。在吉非替尼治疗期间,通过缬沙坦阻断1型血管紧张素II受体(AT1R)可保护心脏,并使心脏标志物、AngII水平及其受体的表达恢复正常。缬沙坦减轻了吉非替尼诱导的NADPH氧化酶和氧化应激,导致JNK/p38-MAPK途径下调。总体而言,AT1R阻断可调节AngII诱导的NADPH氧化酶和JNK/p38-MAPK,从而减轻吉非替尼诱导的心脏肥大。本研究发现AngII/AT1R信号在吉非替尼诱导的心脏肥大中起关键作用,这可能为表皮生长因子受体抑制剂(EGFRIs)诱导的心脏毒性管理提供新方法。