Human Genetics Laboratory, National Institute of Genetics, Mishima 411-8540, Japan.
Laboratory of Computational Genomics, Tokyo University of Pharmacy and Life Sciences, Hachioji 192-0392, Japan.
Int J Mol Sci. 2022 Sep 22;23(19):11124. doi: 10.3390/ijms231911124.
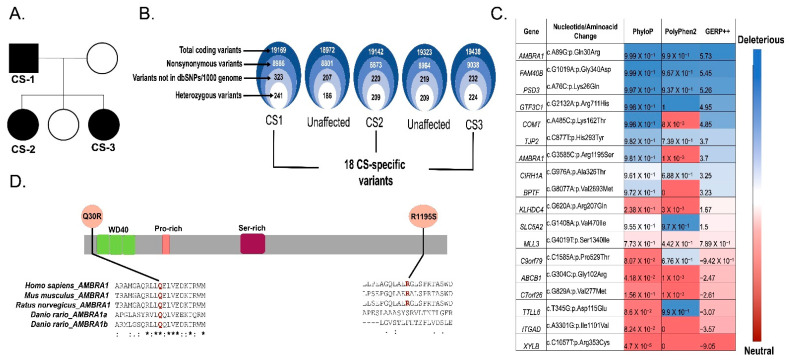
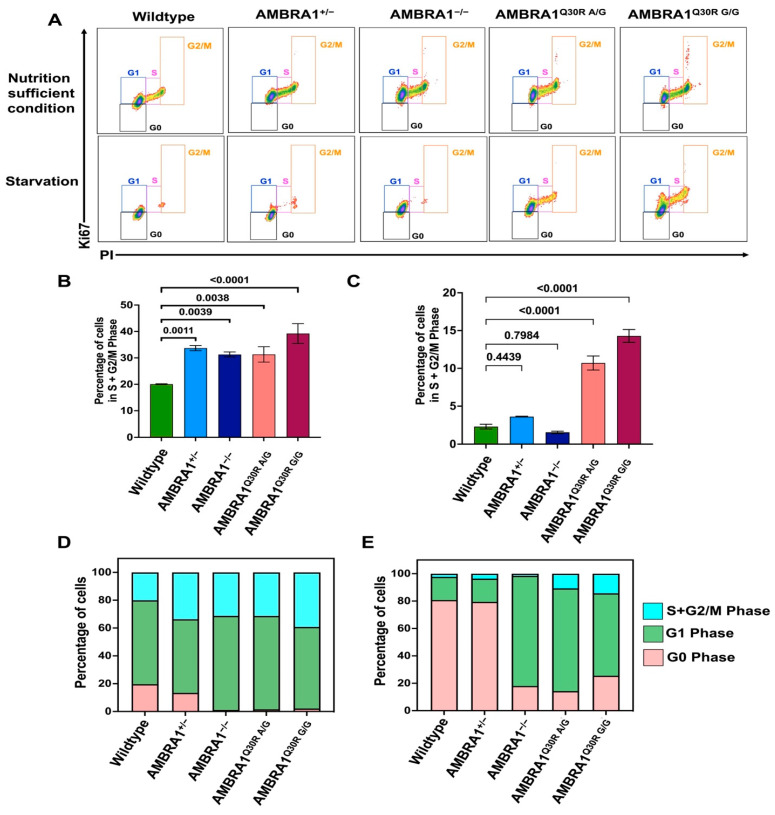
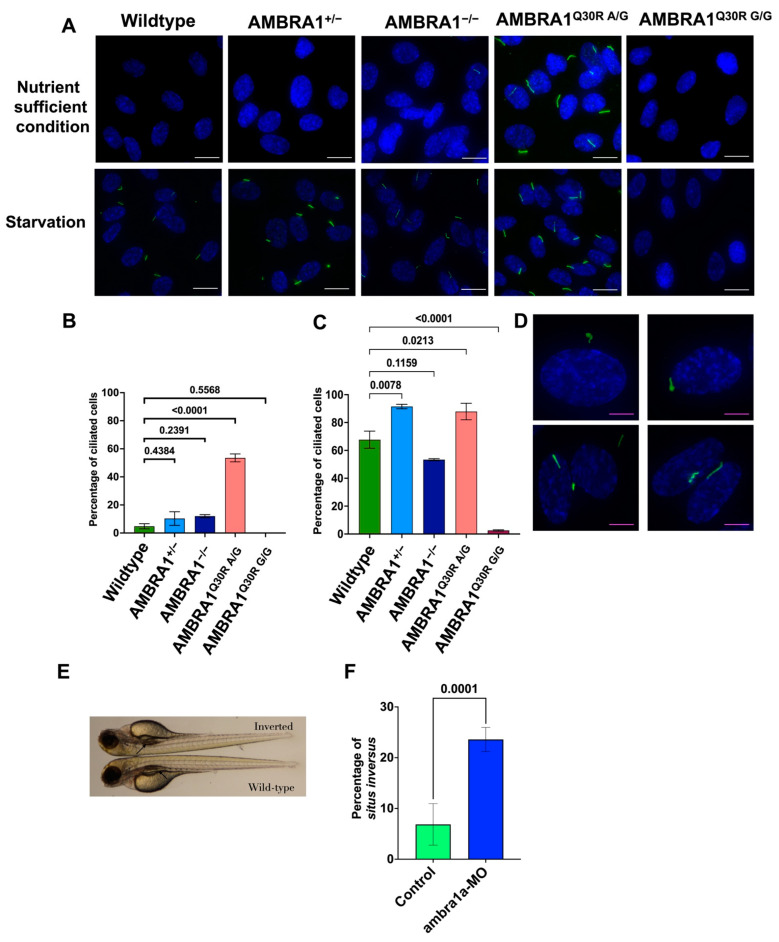
Cowden syndrome (CS) is a rare autosomal dominant disorder associated with multiple hamartomatous and neoplastic lesions in various organs. Most CS patients have been found to have germline mutations in the tumor suppressor. In the present study, we investigated the causative gene of CS in a family of (phosphatase and tensin homolog deleted on chromosome 10) -negative CS patients. Whole exome sequencing analysis revealed (Autophagy and Beclin 1 Regulator 1) as a novel candidate gene harboring two germline variants: p.Gln30Arg (Q30R) and p.Arg1195Ser (R1195S). is a key regulator of the autophagy signaling network and a tumor suppressor. To functionally validate the role of in the clinical manifestations of CS, we generated depletion and Q30R mutation in hTERT-RPE1 (humanTelomerase Reverse Transcriptase-immortalized Retinal Pigmented Epithelial cells) using the CRISPR-Cas9 gene editing system. We observed that both -depleted and mutant cells showed accumulation in the S phase, leading to hyperproliferation, which is a characteristic of hamartomatous lesions. Specifically, the Q30R mutation disturbed the G1/S transition of cells, leading to continuous mitotic entry of mutant cells, irrespective of the extracellular condition. From our analysis of primary ciliogenesis in these cells, we speculated that the mitotic entry of Q30R mutants could be due to non-functional primary cilia that lead to impaired processing of extracellular sensory signals. Additionally, we observed a phenotype in -depleted zebrafish, a developmental abnormality resulting from dysregulated primary ciliogenesis. Taken together, we established that the Q30R mutation that we observed in CS patients might play an important role in inducing the hyperproliferative potential of cells through regulating primary ciliogenesis.
考登综合征(CS)是一种罕见的常染色体显性遗传疾病,与多种器官的错构瘤和肿瘤病变有关。大多数 CS 患者已被发现存在肿瘤抑制基因的种系突变。在本研究中,我们对一组 PSEN1(phosphatase and tensin homolog deleted on chromosome 10)阴性 CS 患者的 CS 致病基因进行了研究。全外显子组测序分析显示,AUTS2(Autophagy and Beclin 1 Regulator 1)是一种新的候选基因,携带两种种系变异:p.Gln30Arg(Q30R)和 p.Arg1195Ser(R1195S)。AUTS2 是自噬信号网络的关键调节因子,也是一种肿瘤抑制因子。为了从功能上验证 AUTS2 在 CS 临床表现中的作用,我们使用 CRISPR-Cas9 基因编辑系统在 hTERT-RPE1(humanTelomerase Reverse Transcriptase-immortalized Retinal Pigmented Epithelial cells)中构建了 AUTS2 敲除和 Q30R 突变。我们观察到,AUTS2 敲除和突变细胞都在 S 期积累,导致过度增殖,这是错构瘤病变的特征。具体来说,AUTS2 的 Q30R 突变干扰了细胞的 G1/S 转换,导致突变细胞持续进入有丝分裂,而不管细胞外环境如何。从我们对这些细胞的初级纤毛发生的分析中,我们推测,AUTS2 Q30R 突变体的有丝分裂进入可能是由于功能失调的初级纤毛,导致细胞外感觉信号处理受损。此外,我们还观察到 AUTS2 敲除斑马鱼出现 表型,这是一种由于初级纤毛发生失调导致的发育异常。综上所述,我们发现 CS 患者中观察到的 AUTS2 Q30R 突变可能通过调节初级纤毛发生在诱导细胞过度增殖潜能方面发挥重要作用。