Department of Gynecologic Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China.
Department of Gastrointestinal Oncology, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Peking University Cancer Hospital & Institute, Beijing, China.
BMC Gastroenterol. 2022 Oct 14;22(1):435. doi: 10.1186/s12876-022-02510-8.
Stomach adenocarcinoma (STAD) is a highly heterogeneous disease and is among the leading causes of cancer-related death worldwide. At present, TNM stage remains the most effective prognostic factor for STAD. Exploring the changes in gene expression levels associated with TNM stage development may help oncologists to better understand the commonalities in the progression of STAD and may provide a new way of identifying early-stage STAD so that optimal treatment approaches can be provided.
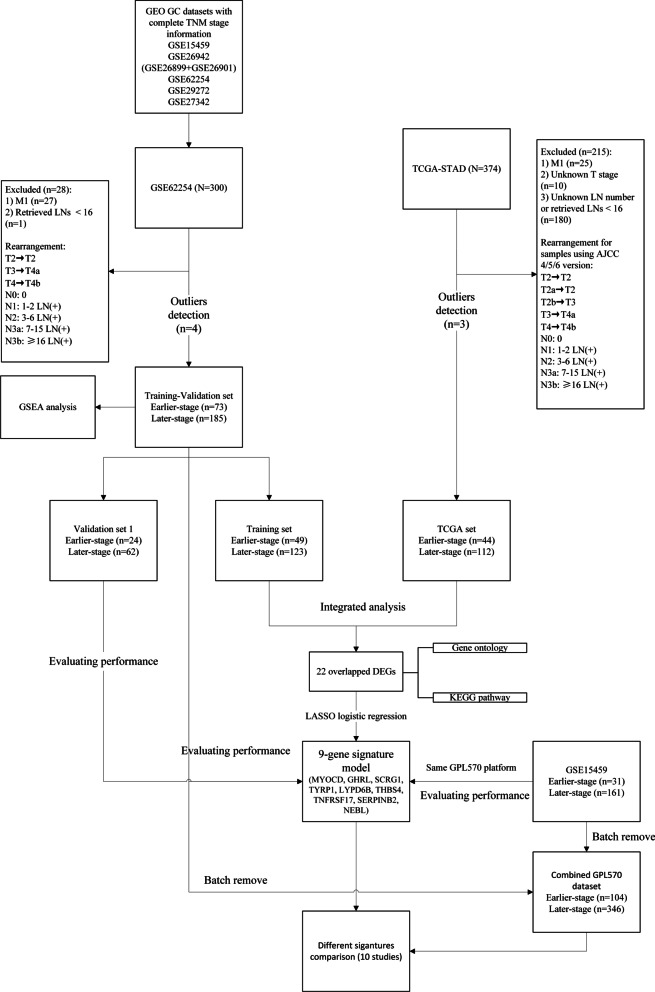
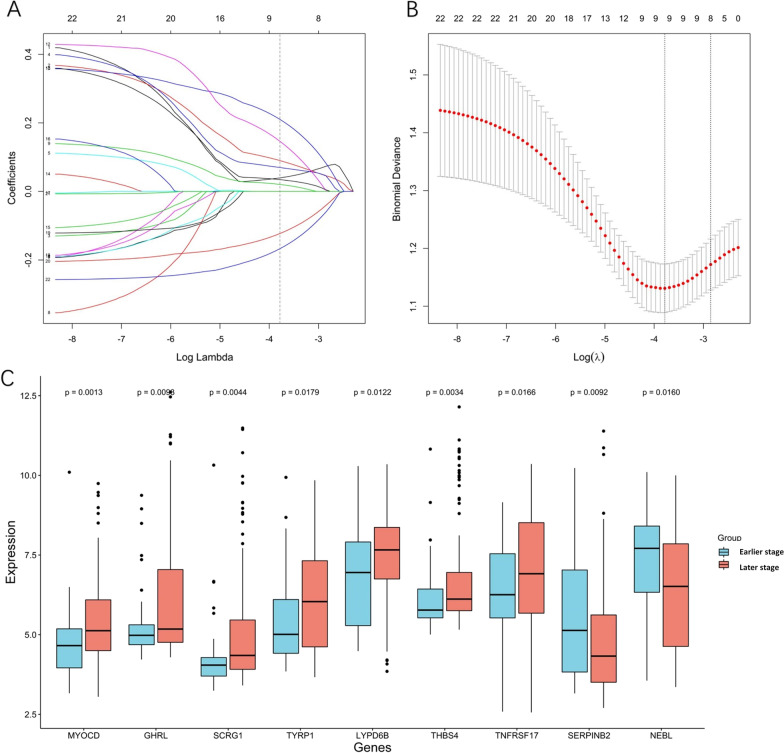
The RNA profile retrieving strategy was utilized and RNA expression profiling was performed using two large STAD microarray databases (GSE62254, n = 300; GSE15459, n = 192) from the Gene Expression Omnibus (GEO) and the RNA-seq database within the Cancer Genome Atlas (TCGA, n = 375). All sample expression information was obtained from STAD tissues after radical resection. After excluding data with insufficient staging information and lymph node number, samples were grouped into earlier-stage and later-stage. Samples in GSE62254 were randomly divided into a training group (n = 172) and a validation group (n = 86). Differentially expressed genes (DEGs) were selected based on the expression of mRNAs in the training group and the TCGA group (n = 156), and hub genes were further screened by least absolute shrinkage and selection operator (LASSO) logistic regression. Receiver operating characteristic (ROC) curves were used to evaluate the performance of the hub genes in distinguishing STAD stage in the validation group and the GSE15459 dataset. Univariate and multivariate Cox regressions were performed sequentially.
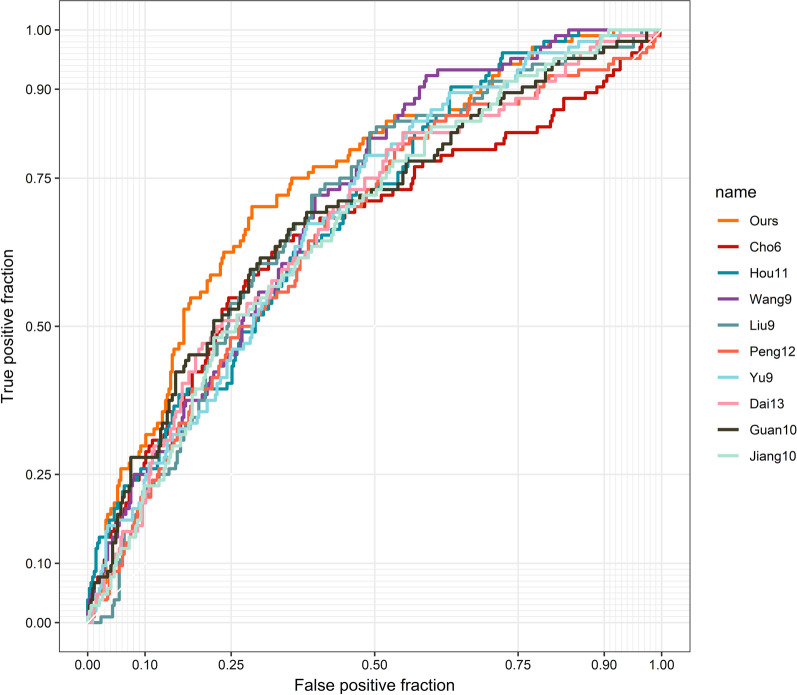
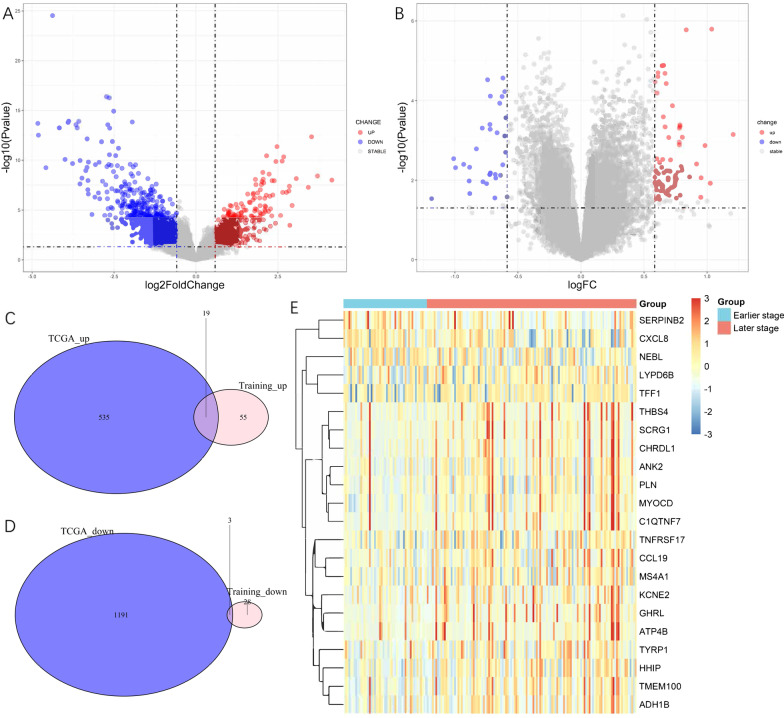
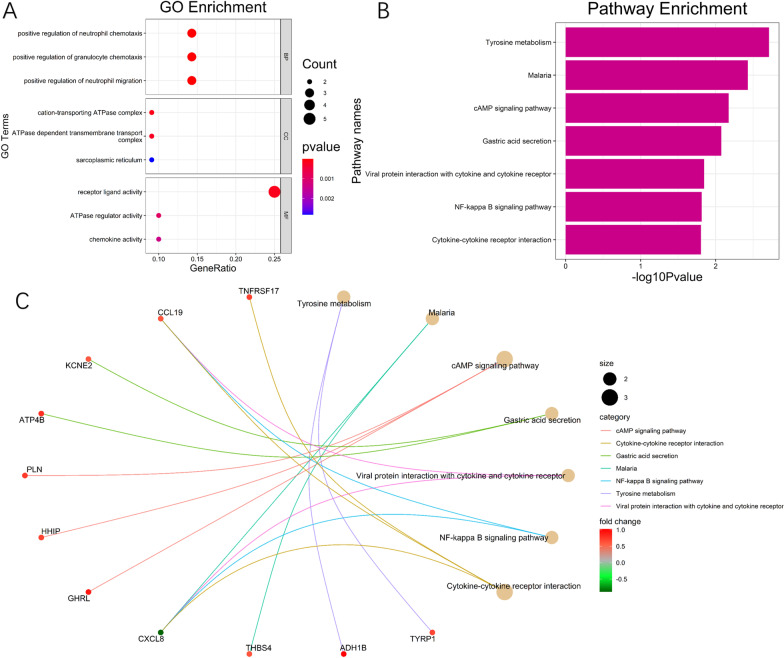
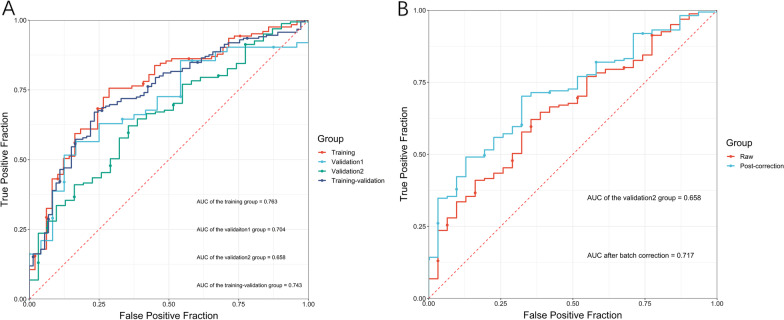
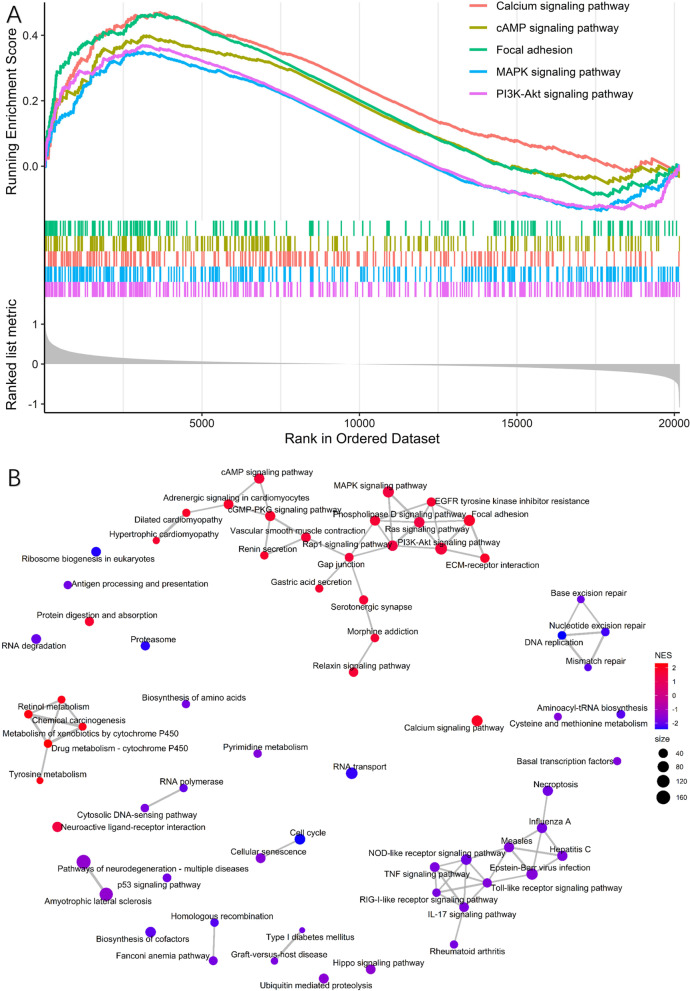
22 DEGs were commonly upregulated (n = 19) or downregulated (n = 3) in the training and TCGA datasets. Nine genes, including MYOCD, GHRL, SCRG1, TYRP1, LYPD6B, THBS4, TNFRSF17, SERPINB2, and NEBL were identified as hub genes by LASSO-logistic regression. The model achieved discrimination in the validation group (AUC = 0.704), training-validation group (AUC = 0.743), and GSE15459 dataset (AUC = 0.658), respectively. Gene Set Enrichment Analysis (GSEA) was used to identify the potential stage-development pathways, including the PI3K-Akt and Calcium signaling pathways. Univariate Cox regression indicated that the nine-gene score was a significant risk factor for overall survival (HR = 1.28, 95% CI 1.08-1.50, P = 0.003). In the multivariate Cox regression, only SCRG1 was an independent prognostic predictor of overall survival after backward stepwise elimination (HR = 1.21, 95% CI 1.11-1.32, P < 0.001).
Through a series of bioinformatics and validation processes, a nine-gene signature that can distinguish STAD stage was identified. This gene signature has potential clinical application and may provide a novel approach to understanding the progression of STAD.
胃腺癌(STAD)是一种高度异质性疾病,也是全球癌症相关死亡的主要原因之一。目前,TNM 分期仍然是 STAD 最有效的预后因素。探索与 TNM 分期发展相关的基因表达水平变化,可能有助于肿瘤学家更好地理解 STAD 进展的共同特征,并可能提供一种识别早期 STAD 的新方法,以便提供最佳治疗方法。
利用 RNA 谱检索策略,使用来自基因表达综合数据库(GEO)的两个大型 STAD 微阵列数据库(GSE62254,n=300;GSE15459,n=192)和癌症基因组图谱(TCGA)中的 RNA-seq 数据库(n=375)进行 RNA 表达谱分析。所有样本的表达信息均来自根治性切除后的 STAD 组织。在排除分期信息和淋巴结数量不足的数据后,将样本分为早期和晚期。GSE62254 中的样本被随机分为训练组(n=172)和验证组(n=86)。基于训练组和 TCGA 组(n=156)中 mRNA 的表达选择差异表达基因(DEGs),并通过最小绝对收缩和选择算子(LASSO)逻辑回归进一步筛选枢纽基因。接收器工作特征(ROC)曲线用于评估枢纽基因在验证组和 GSE15459 数据集区分 STAD 分期的性能。依次进行单变量和多变量 Cox 回归。
在训练组和 TCGA 数据集中共发现 22 个上调(n=19)或下调(n=3)的 DEG。通过 LASSO-逻辑回归鉴定出 9 个基因(包括 MYOCD、GHRL、SCRG1、TYRP1、LYPD6B、THBS4、TNFRSF17、SERPINB2 和 NEBL)为枢纽基因。该模型在验证组(AUC=0.704)、训练-验证组(AUC=0.743)和 GSE15459 数据集(AUC=0.658)中均实现了区分。基因集富集分析(GSEA)用于鉴定潜在的分期发展途径,包括 PI3K-Akt 和钙信号通路。单变量 Cox 回归表明,九个基因的评分是总生存期的显著危险因素(HR=1.28,95%CI 1.08-1.50,P=0.003)。在多变量 Cox 回归中,只有 SCRG1 是总生存期的独立预后预测因子(HR=1.21,95%CI 1.11-1.32,P<0.001)。
通过一系列生物信息学和验证过程,确定了一个可以区分 STAD 分期的九个基因特征。该基因特征具有潜在的临床应用价值,并可能为理解 STAD 的进展提供一种新方法。