Laboratorio di Biologia Bio@SNS, Scuola Normale Superiore, Pisa, Italy.
University of Siena, DIISM-SAILAB, Siena, Italy.
Commun Biol. 2022 Oct 26;5(1):1133. doi: 10.1038/s42003-022-04073-6.
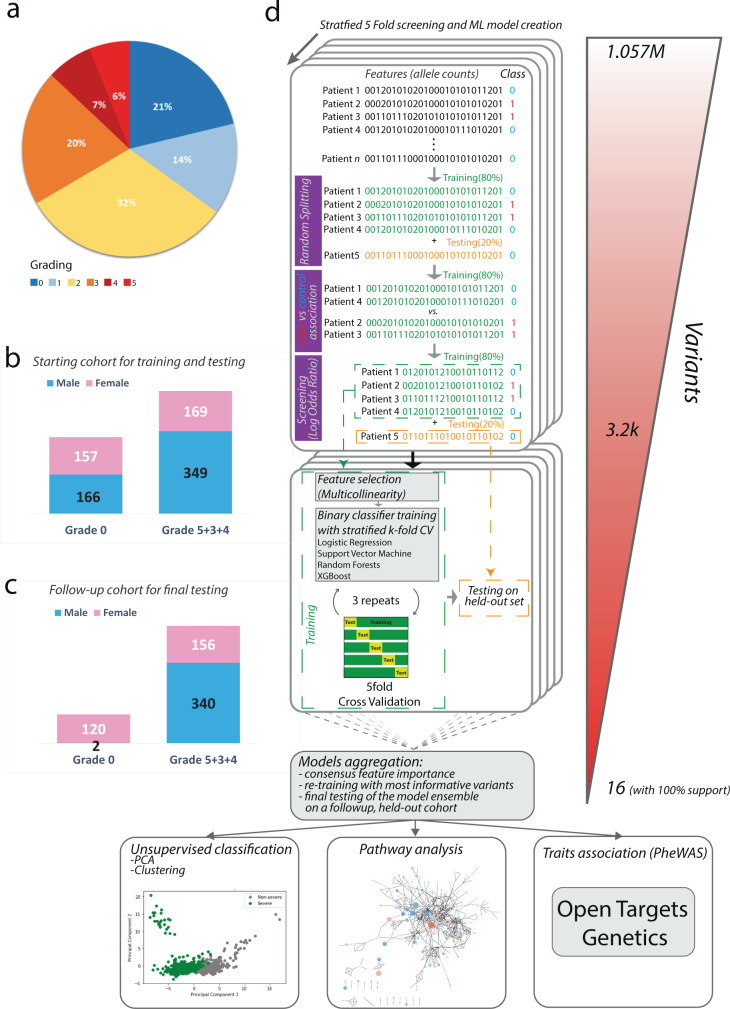
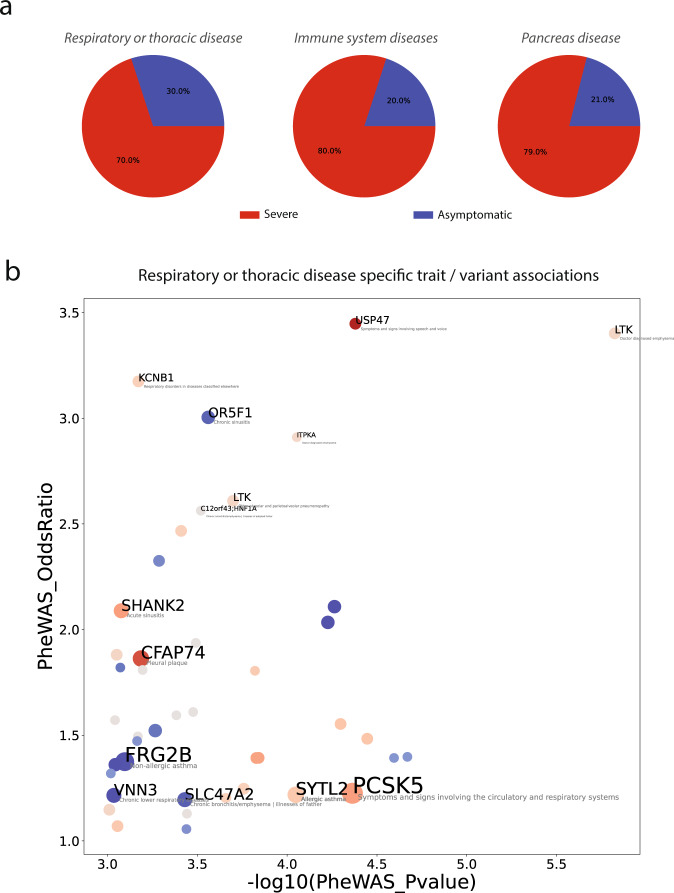
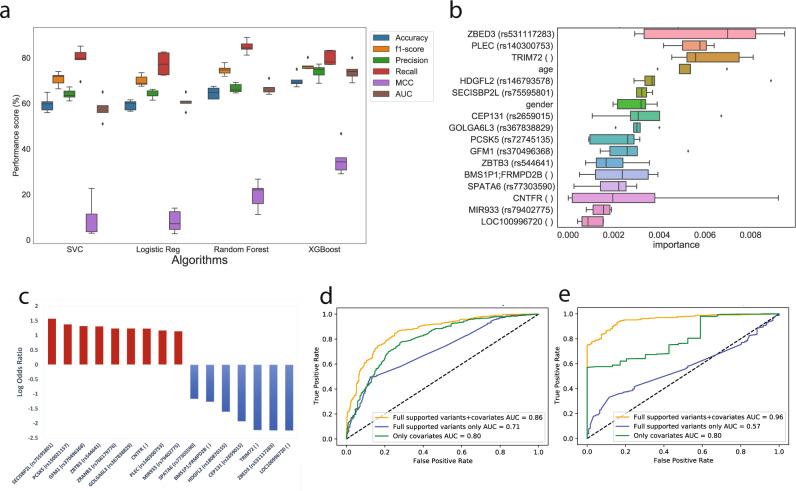
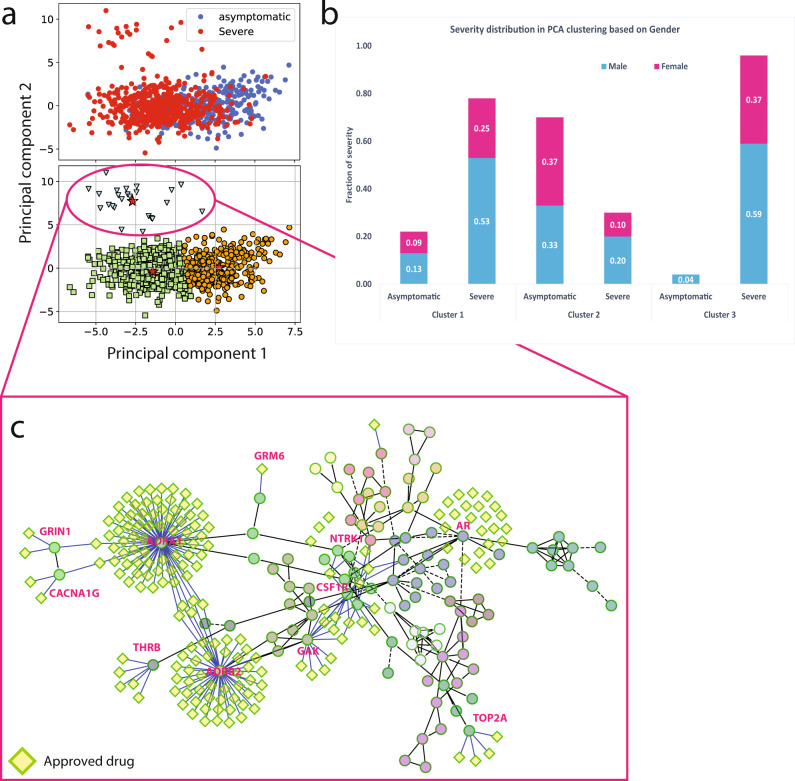
We employed a multifaceted computational strategy to identify the genetic factors contributing to increased risk of severe COVID-19 infection from a Whole Exome Sequencing (WES) dataset of a cohort of 2000 Italian patients. We coupled a stratified k-fold screening, to rank variants more associated with severity, with the training of multiple supervised classifiers, to predict severity based on screened features. Feature importance analysis from tree-based models allowed us to identify 16 variants with the highest support which, together with age and gender covariates, were found to be most predictive of COVID-19 severity. When tested on a follow-up cohort, our ensemble of models predicted severity with high accuracy (ACC = 81.88%; AUCROC = 96%; MCC = 61.55%). Our model recapitulated a vast literature of emerging molecular mechanisms and genetic factors linked to COVID-19 response and extends previous landmark Genome-Wide Association Studies (GWAS). It revealed a network of interplaying genetic signatures converging on established immune system and inflammatory processes linked to viral infection response. It also identified additional processes cross-talking with immune pathways, such as GPCR signaling, which might offer additional opportunities for therapeutic intervention and patient stratification. Publicly available PheWAS datasets revealed that several variants were significantly associated with phenotypic traits such as "Respiratory or thoracic disease", supporting their link with COVID-19 severity outcome.
我们采用了一种多方面的计算策略,从一个由 2000 名意大利患者组成的队列的全外显子组测序 (WES) 数据集中,确定导致 COVID-19 严重感染风险增加的遗传因素。我们将分层 k 折筛选与多种监督分类器的训练相结合,根据筛选的特征预测严重程度。基于树的模型的特征重要性分析使我们能够识别出 16 个具有最高支持率的变体,这些变体与年龄和性别协变量一起,被发现是 COVID-19 严重程度的最具预测性因素。当在后续队列中进行测试时,我们的模型集合能够以高精度预测严重程度 (ACC=81.88%; AUCROC=96%; MCC=61.55%)。我们的模型再现了大量与 COVID-19 反应相关的新兴分子机制和遗传因素的文献,并扩展了以前的标志性全基因组关联研究 (GWAS)。它揭示了一个相互作用的遗传特征网络,这些特征集中在与病毒感染反应相关的已建立的免疫系统和炎症过程上。它还确定了与免疫途径相互作用的其他过程,例如 GPCR 信号转导,这可能为治疗干预和患者分层提供额外的机会。公开可用的 PheWAS 数据集表明,几个变体与“呼吸或胸部疾病”等表型特征显著相关,支持它们与 COVID-19 严重程度结果的联系。