Department of Endocrinology, Translational Research Key Laboratory for Diabetes, The Second Affiliated Hospital, Xinqiao Hospital, Army Medical University, Chongqing, China.
Department of Medicine, Division of Diabetes, Endocrinology and Metabolism, Baylor College of Medicine, Houston, TX, United States.
Front Endocrinol (Lausanne). 2022 Oct 19;13:989447. doi: 10.3389/fendo.2022.989447. eCollection 2022.
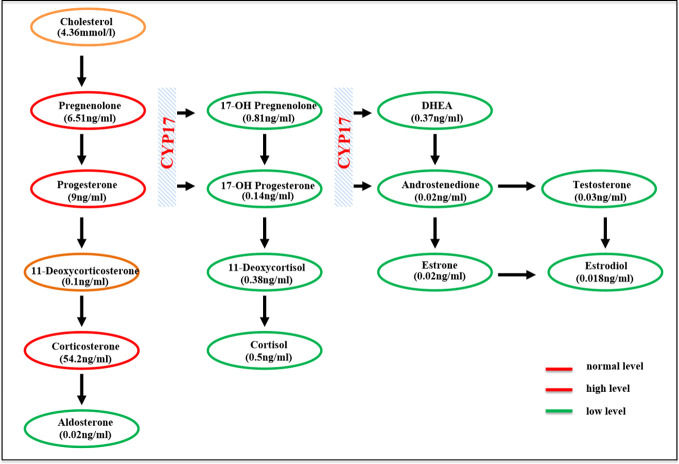
Combined 17α-hydroxylase/17,20-lyase deficiency (17-OHD) is a very rare form of congenital adrenal hyperplasia (CAH) caused by mutations in the CYP17A1 gene. Almost 100 different mutations of the CYP17A1 gene have been reported, including p.R96Q mutation, but no case of p.R96Q mutation has been described in Asian populations.
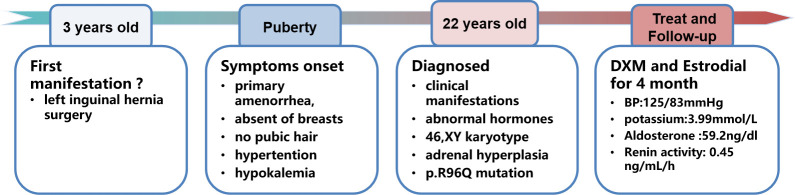
We describe a 22-year-old female patient of 46,XY karyotype, who presented with pseudohermaphrodism, primary amenorrhea, underdeveloped secondary sexual characteristics, delayed epiphyseal healing, hypertension, and hypokalemia. The diagnosis of 17-OHD was reached by measurement of steroid hormones and abdominal CT scan and confirmed by genetic sequencing, which revealed a homozygous p.R96Q missense mutation in the CYP17A1 gene. The patient received treatment with dexamethasone and estradiol, and 4 months of follow-up showed that both blood pressure and potassium were well controlled.
This is the first Asian case of CAH caused by a homozygous p.R96Q missense mutation in the CYP17A1 gene. Herein, we highlight the role of inguinal hernia in the early diagnosis of female 17-OHD and the necessity of removing the ectopic testis.
17α-羟化酶/17,20-裂合酶缺陷(17-OHD)是一种非常罕见的先天性肾上腺增生症(CAH)形式,由 CYP17A1 基因突变引起。已经报道了 CYP17A1 基因的近 100 种不同突变,包括 p.R96Q 突变,但在亚洲人群中尚未描述过 p.R96Q 突变的病例。
我们描述了一名 22 岁的 46,XY 核型女性患者,表现为假两性畸形、原发性闭经、第二性征发育不良、骨骺愈合延迟、高血压和低钾血症。17-OHD 的诊断通过类固醇激素和腹部 CT 扫描测量得出,并通过基因测序证实,该患者 CYP17A1 基因存在纯合 p.R96Q 错义突变。患者接受了地塞米松和雌二醇治疗,4 个月的随访显示血压和血钾均得到良好控制。
这是首例 CYP17A1 基因纯合 p.R96Q 错义突变导致的 CAH 亚洲病例。本文强调了腹股沟疝在女性 17-OHD 早期诊断中的作用,以及切除异位睾丸的必要性。