School of Medicine, The Stanley Scott Cancer Center/Louisiana Cancer Research Center, Louisiana State University Health Sciences Center, New Orleans, LA, USA.
Department of Microbiology and Immunology, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt.
J Transl Med. 2022 Nov 8;20(1):521. doi: 10.1186/s12967-022-03715-x.
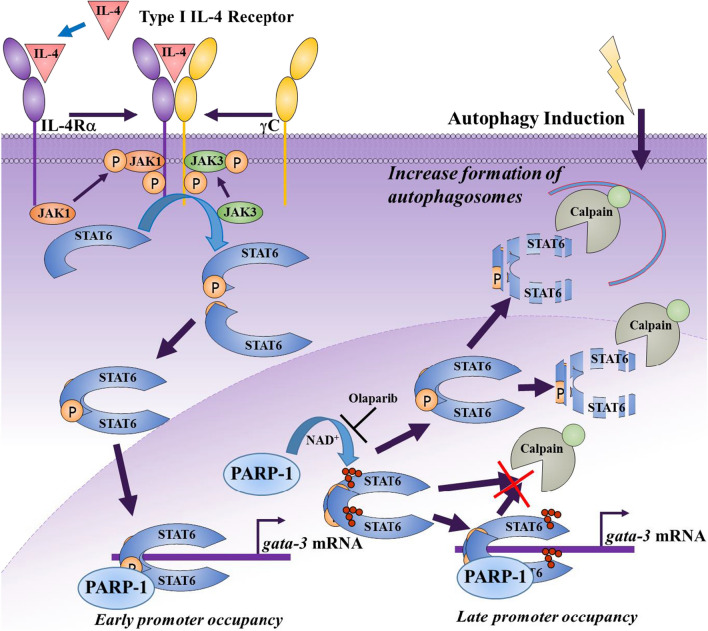
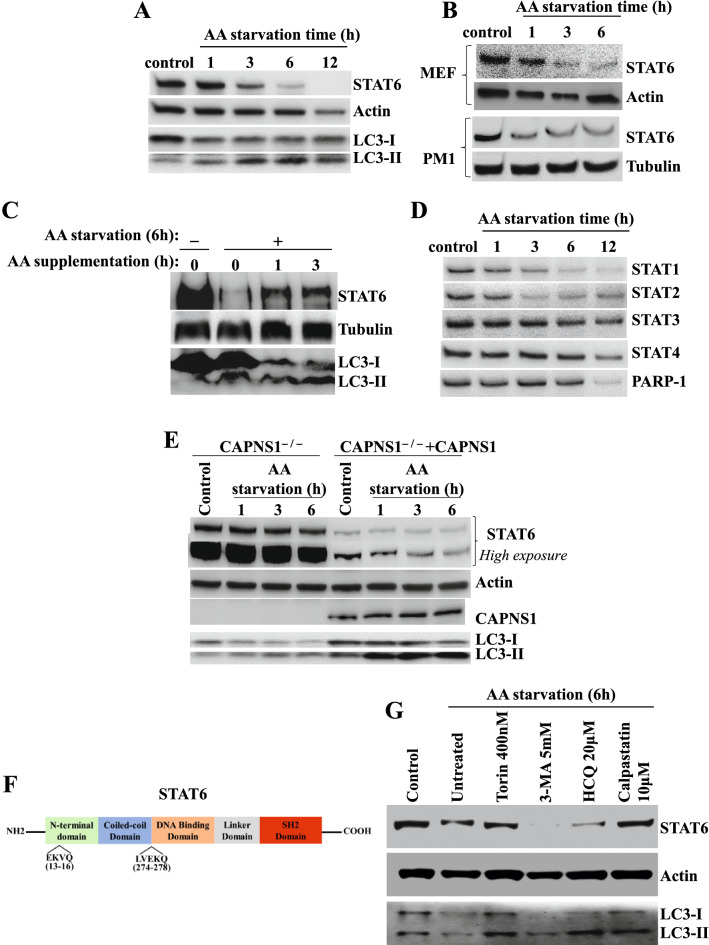
We reported that PARP-1 regulates genes whose products are crucial for asthma, in part, by controlling STAT6 integrity speculatively through a calpain-dependent mechanism. We wished to decipher the PARP-1/STAT6 relationship in the context of intracellular trafficking and promoter occupancy of the transcription factor on target genes, its integrity in the presence of calpains, and its connection to autophagy.
This study was conducted using primary splenocytes or fibroblasts derived from wild-type or PARP-1 mice and Jurkat T cells to mimic Th2 inflammation.
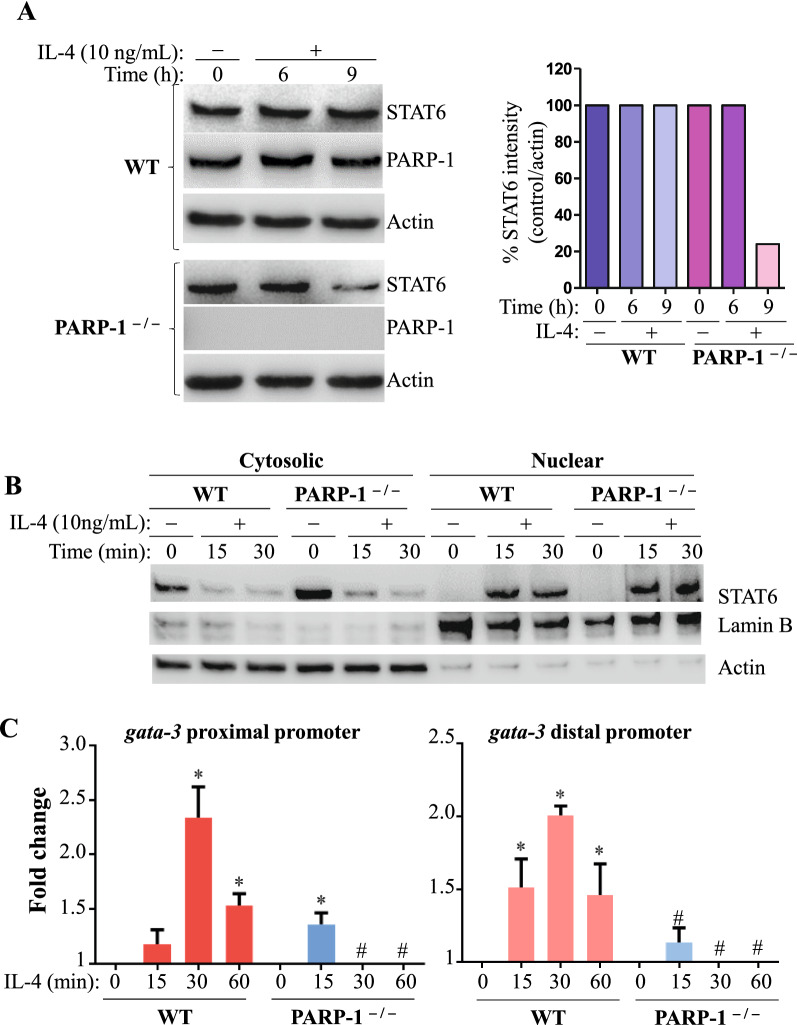
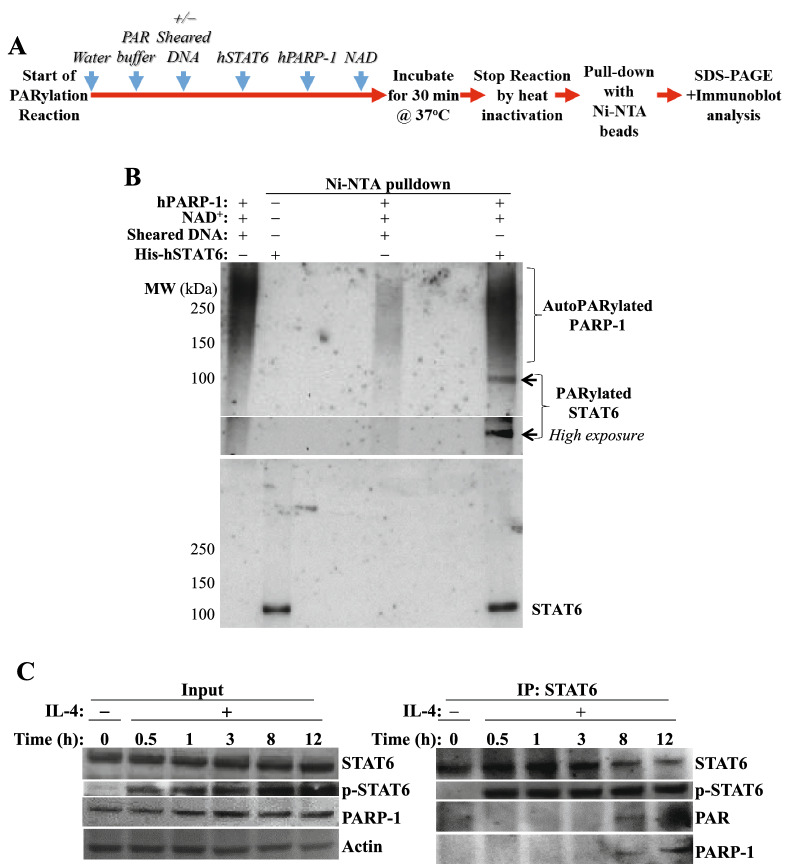
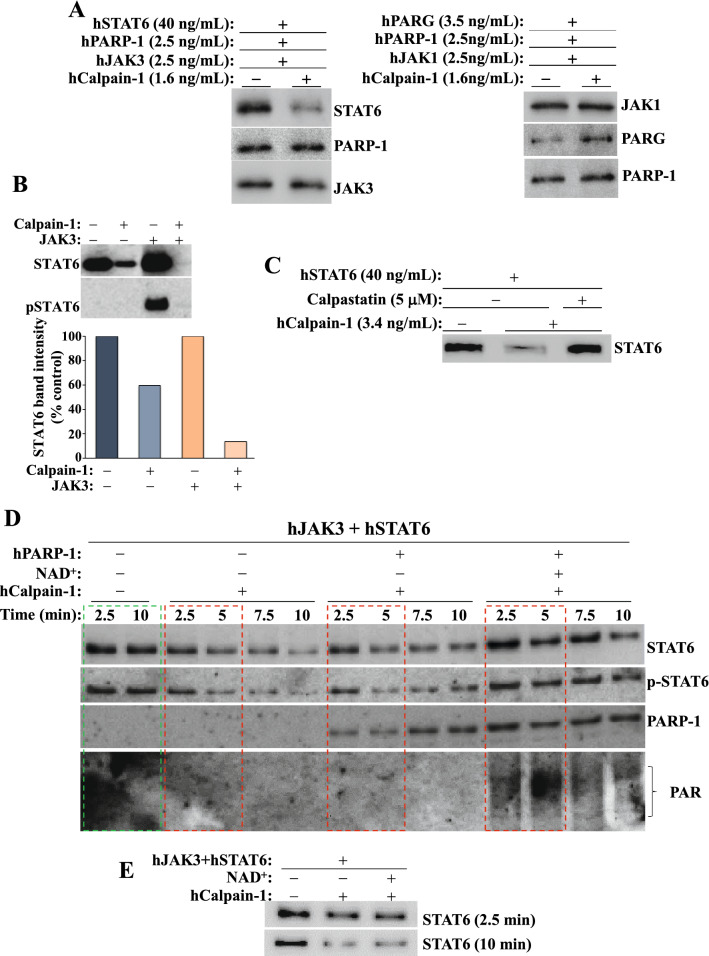
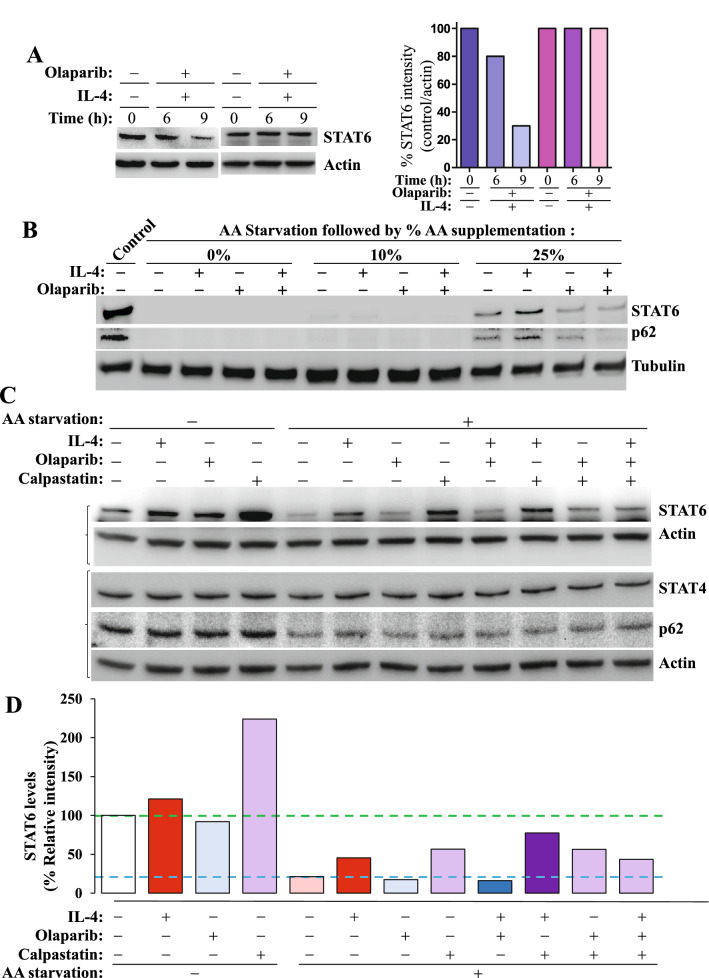
We show that the role for PARP-1 in expression of IL-4-induced genes (e.g. gata-3) in splenocytes did not involve effects on STAT6 phosphorylation or its subcellular trafficking, rather, it influenced its occupancy of gata-3 proximal and distal promoters in the early stages of IL-4 stimulation. At later stages, PARP-1 was crucial for STAT6 integrity as its inhibition, pharmacologically or by gene knockout, compromised the fate of the transcription factor. Calpain-1 appeared to preferentially degrade JAK-phosphorylated-STAT6, which was blocked by calpastatin-mediated inhibition or by genetic knockout in mouse fibroblasts. The STAT6/PARP-1 relationship entailed physical interaction and modification by poly(ADP-ribosyl)ation independently of double-strand-DNA breaks. Poly(ADP-ribosyl)ation protected phosphorylated-STAT6 against calpain-1-mediated degradation. Additionally, our results show that STAT6 is a bonafide substrate for chaperone-mediated autophagy in a selective and calpain-dependent manner in the human Jurkat cell-line. The effects were partially blocked by IL-4 treatment and PARP-1 inhibition.
The results demonstrate that poly(ADP-ribosyl)ation plays a critical role in protecting activated STAT6 during Th2 inflammation, which may be synthetically targeted for degradation by inhibiting PARP-1.
我们曾报道过,PARP-1 通过一种钙蛋白酶依赖性机制,推测性地控制 STAT6 的完整性,从而调节对哮喘至关重要的基因产物。我们希望在细胞内运输和转录因子对靶基因启动子的占据、钙蛋白酶存在下其完整性及其与自噬的关系的背景下,解析 PARP-1/STAT6 的关系。
本研究使用源自野生型或 PARP-1 小鼠的原代脾细胞或成纤维细胞和 Jurkat T 细胞来模拟 Th2 炎症。
我们表明,PARP-1 在脾细胞中诱导的 IL-4 基因(例如 gata-3)表达中的作用不涉及 STAT6 磷酸化或其亚细胞运输的影响,而是在 IL-4 刺激的早期影响其对 gata-3 近端和远端启动子的占据。在后期,PARP-1 对 STAT6 的完整性至关重要,因为其抑制作用,无论是药理学还是基因敲除,都会损害转录因子的命运。钙蛋白酶-1 似乎优先降解 JAK 磷酸化的 STAT6,而钙蛋白酶抑制剂或小鼠成纤维细胞中的基因敲除可阻断这一过程。STAT6/PARP-1 关系需要物理相互作用和多聚(ADP-核糖基)化修饰,而不涉及双链 DNA 断裂。多聚(ADP-核糖基)化可保护磷酸化的 STAT6 免受钙蛋白酶-1 介导的降解。此外,我们的结果表明,在人类 Jurkat 细胞系中,STAT6 是伴侣介导自噬的真正底物,具有选择性和钙蛋白酶依赖性。这些影响部分被 IL-4 处理和 PARP-1 抑制所阻断。
结果表明,多聚(ADP-核糖基)化在 Th2 炎症期间保护激活的 STAT6 发挥关键作用,这可能通过抑制 PARP-1 来进行合成靶向降解。