Novo Nordisk Research Center Indianapolis, Indianapolis, IN 46241, USA.
Duke Molecular Physiology Institute, Duke University, Durham, NC, 27705, USA.
Mol Metab. 2022 Dec;66:101638. doi: 10.1016/j.molmet.2022.101638. Epub 2022 Nov 15.
Glucose-dependent insulinotropic polypeptide (GIP) is one of the two major incretin factors that regulate metabolic homeostasis. Genetic ablation of its receptor (GIPR) in mice confers protection against diet-induced obesity (DIO), while GIPR neutralizing antibodies produce additive weight reduction when combined with GLP-1R agonists in preclinical models and clinical trials. Conversely, GIPR agonists have been shown to promote weight loss in rodents, while dual GLP-1R/GIPR agonists have proven superior to GLP-1R monoagonists for weight reduction in clinical trials. We sought to develop a long-acting, specific GIPR peptide antagonist as a tool compound suitable for investigating GIPR pharmacology in both rodent and human systems.
We report a structure-activity relationship of GIPR peptide antagonists based on the human and mouse GIP sequences with fatty acid-based protraction. We assessed these compounds in vitro, in vivo in DIO mice, and ex vivo in islets from human donors.
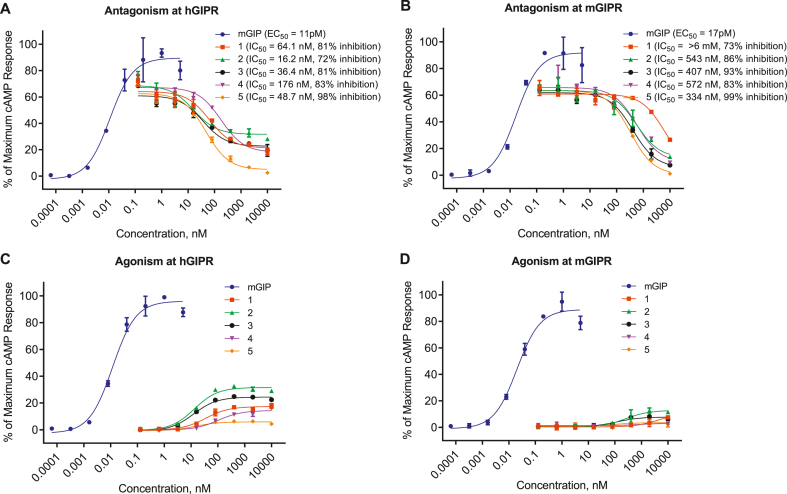
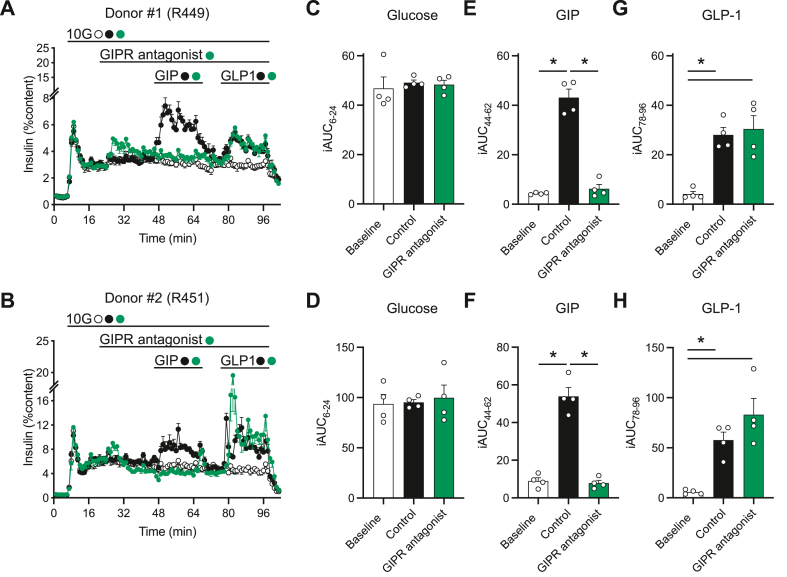
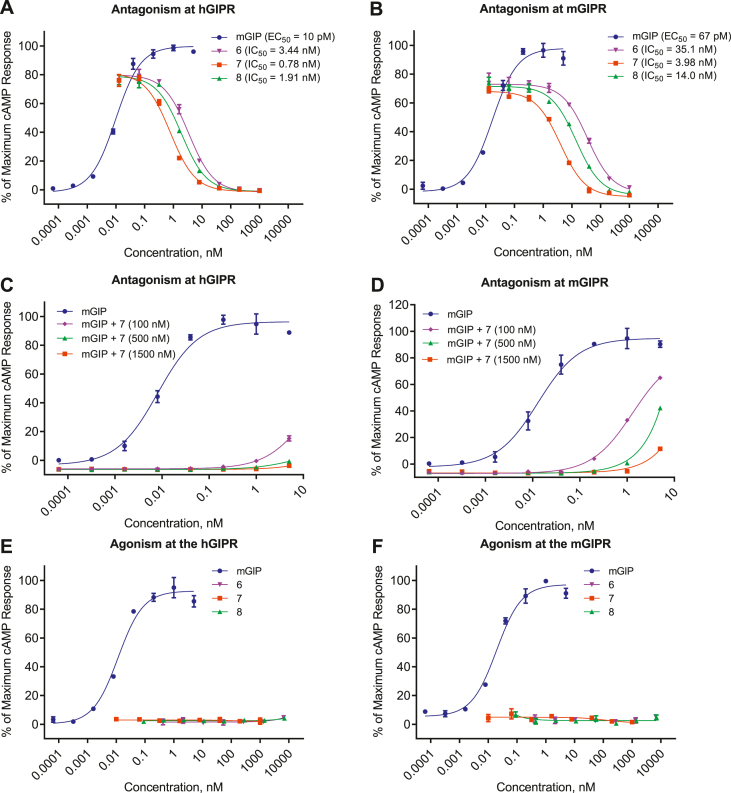
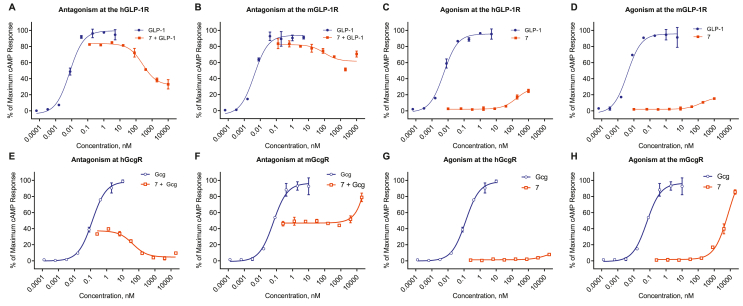
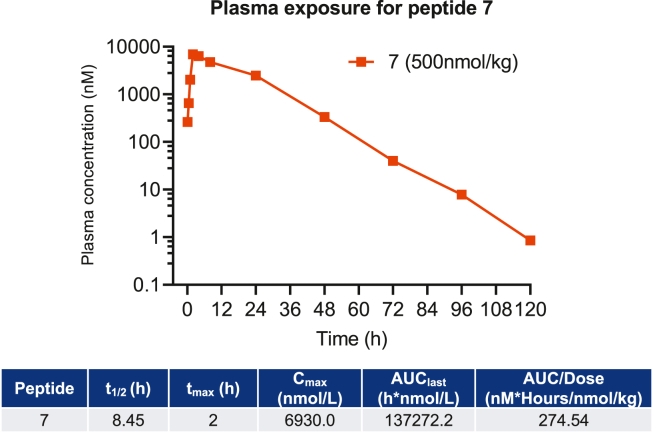
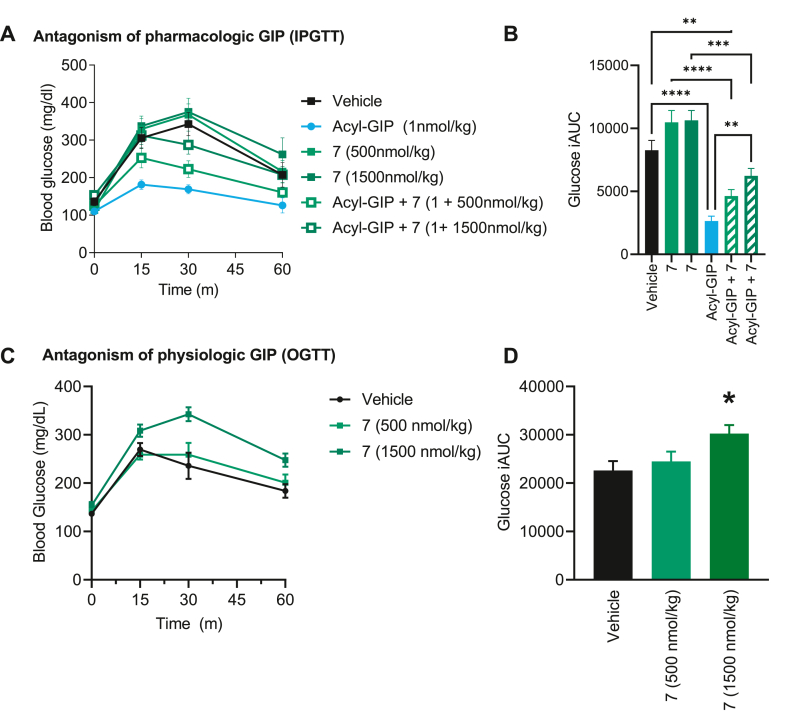
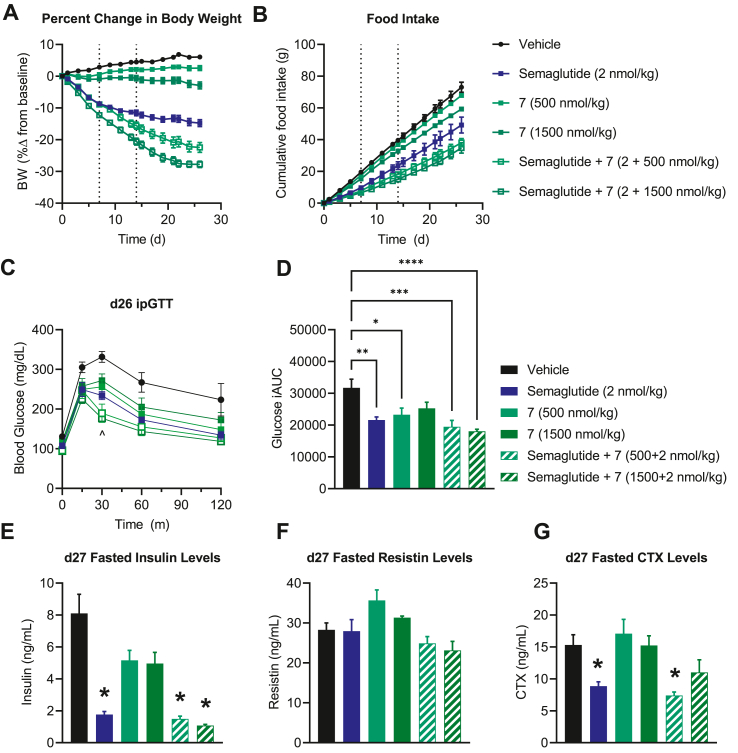
We report the discovery of a GIP palmitoylated analogue, [N-Ac, L14, R18, E21] hGIP-K11 (γE-C16), which potently inhibits in vitro GIP-mediated cAMP generation at both the hGIPR and mGIPR. In vivo, this peptide effectively blocks GIP-mediated reductions in glycemia in response to exogenous and endogenous GIP and displays a circulating pharmacokinetic profile amenable for once-daily dosing in rodents. Co-administration with the GLP-1R agonist semaglutide and this GIPR peptide antagonist potentiates weight loss compared to semaglutide alone. Finally, this antagonist inhibits GIP- but not GLP-1-stimulated insulin secretion in intact human islets.
Our work demonstrates the discovery of a potent, specific, and long-acting GIPR peptide antagonist that effectively blocks GIP action in vitro, ex vivo in human islets, and in vivo in mice while producing additive weight-loss when combined with a GLP-1R agonist in DIO mice.
葡萄糖依赖性胰岛素多肽(GIP)是调节代谢平衡的两种主要肠降血糖素因子之一。在小鼠中敲除其受体(GIPR)可防止饮食诱导的肥胖(DIO),而 GIPR 中和抗体在临床前模型和临床试验中与 GLP-1R 激动剂联合使用可产生附加的体重减轻。相反,GIPR 激动剂已被证明可促进啮齿动物体重减轻,而双重 GLP-1R/GIPR 激动剂在临床试验中已被证明比 GLP-1R 单激动剂更能减轻体重。我们试图开发一种长效、特异性的 GIPR 肽拮抗剂作为一种工具化合物,适合在啮齿动物和人类系统中研究 GIPR 药理学。
我们报告了基于人源和鼠源 GIP 序列的 GIPR 肽拮抗剂的构效关系,并采用脂肪酸基延长。我们在体外、DIO 小鼠体内和人供体胰岛中评估了这些化合物。
我们报告了一种 GIP 棕榈酰化类似物 [N-Ac,L14,R18,E21] hGIP-K11(γE-C16)的发现,该类似物可有效抑制 hGIPR 和 mGIPR 中体外 GIP 介导的 cAMP 生成。在体内,该肽可有效阻断外源性和内源性 GIP 介导的血糖降低,并显示出可用于啮齿动物每日一次给药的循环药代动力学特征。与 GLP-1R 激动剂司美格鲁肽联合使用时,该 GIPR 肽拮抗剂与司美格鲁肽单独使用相比可增强体重减轻。最后,该拮抗剂可抑制完整人胰岛中 GIP-但不 GLP-1 刺激的胰岛素分泌。
我们的工作证明了一种有效的 GIPR 肽拮抗剂的发现,该拮抗剂具有强大、特异性和长效作用,可有效阻断 GIP 在体外、人胰岛中的作用,以及在 DIO 小鼠体内的作用,并且与 GLP-1R 激动剂联合使用时可产生附加的体重减轻。