Institute of Nanotechnology, Karlsruhe Institute of Technology, P.O. Box 3640, 76021, Karlsruhe, Germany.
Helmholtz Institute Ulm (HIU) Electrochemical Energy Storage, Helmholtzstraße 11, Ulm, 89081, Germany.
ChemSusChem. 2023 Feb 8;16(3):e202202090. doi: 10.1002/cssc.202202090. Epub 2023 Jan 13.
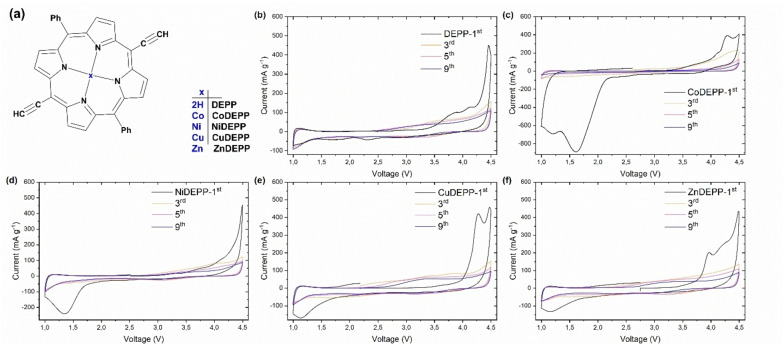
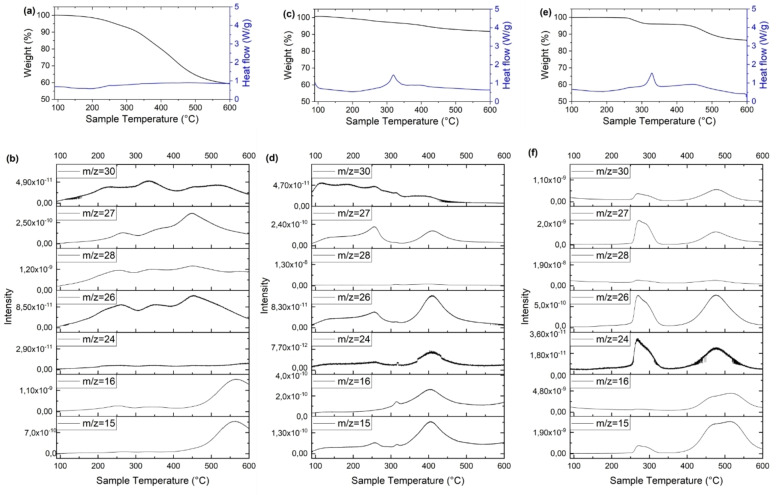
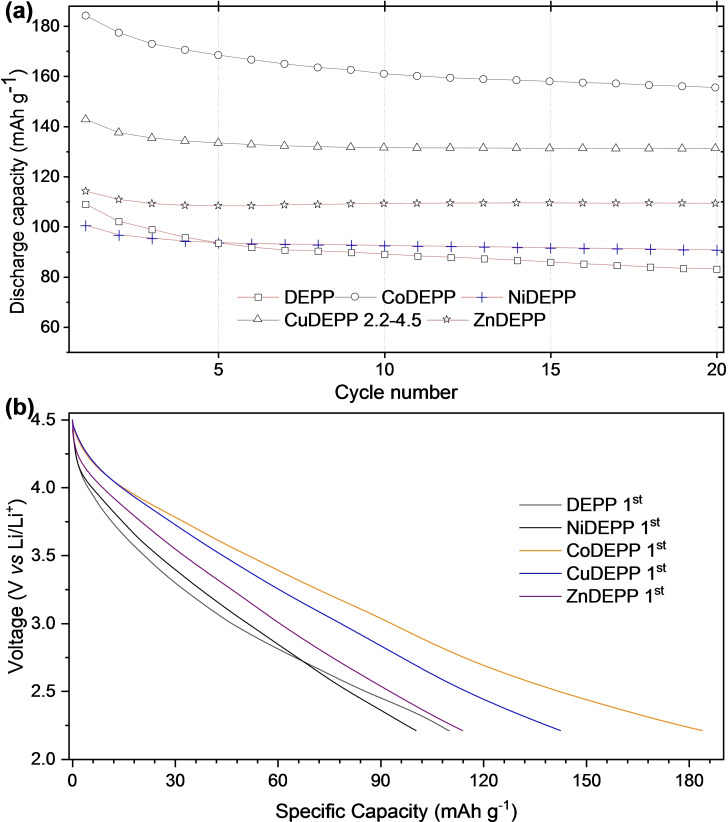
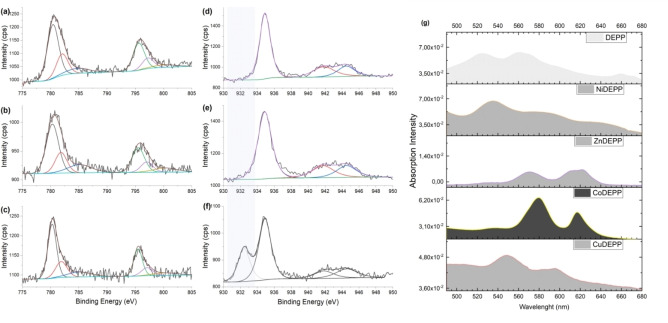
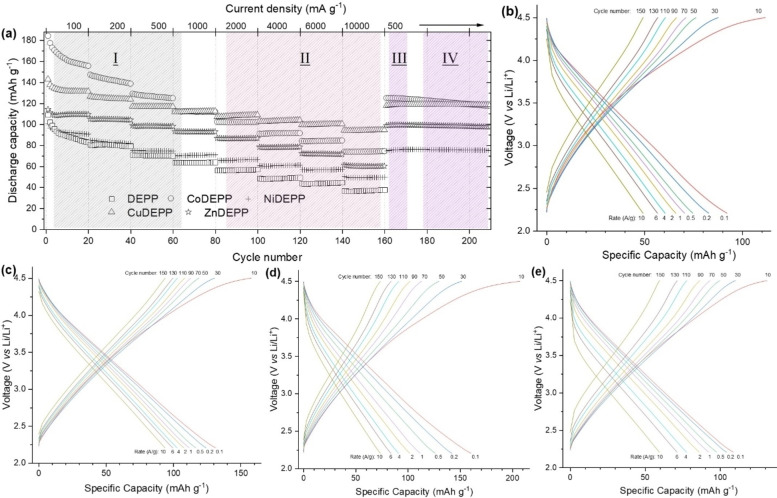
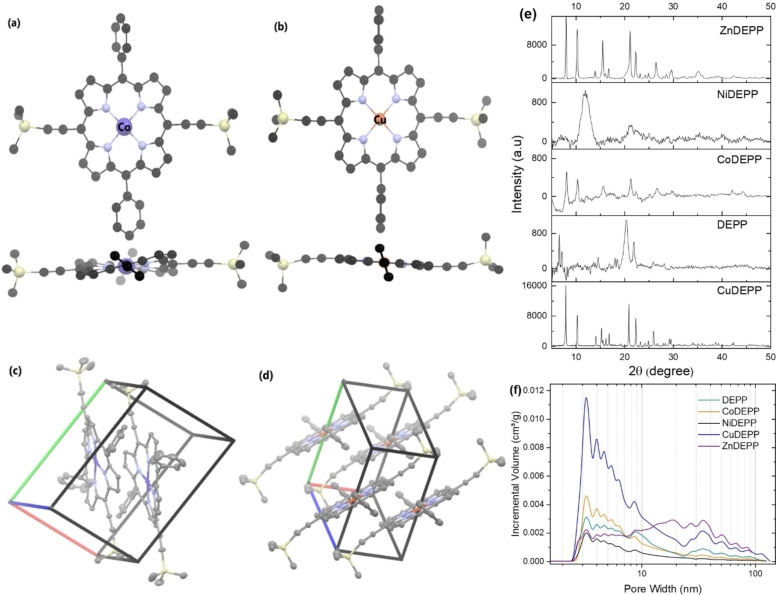
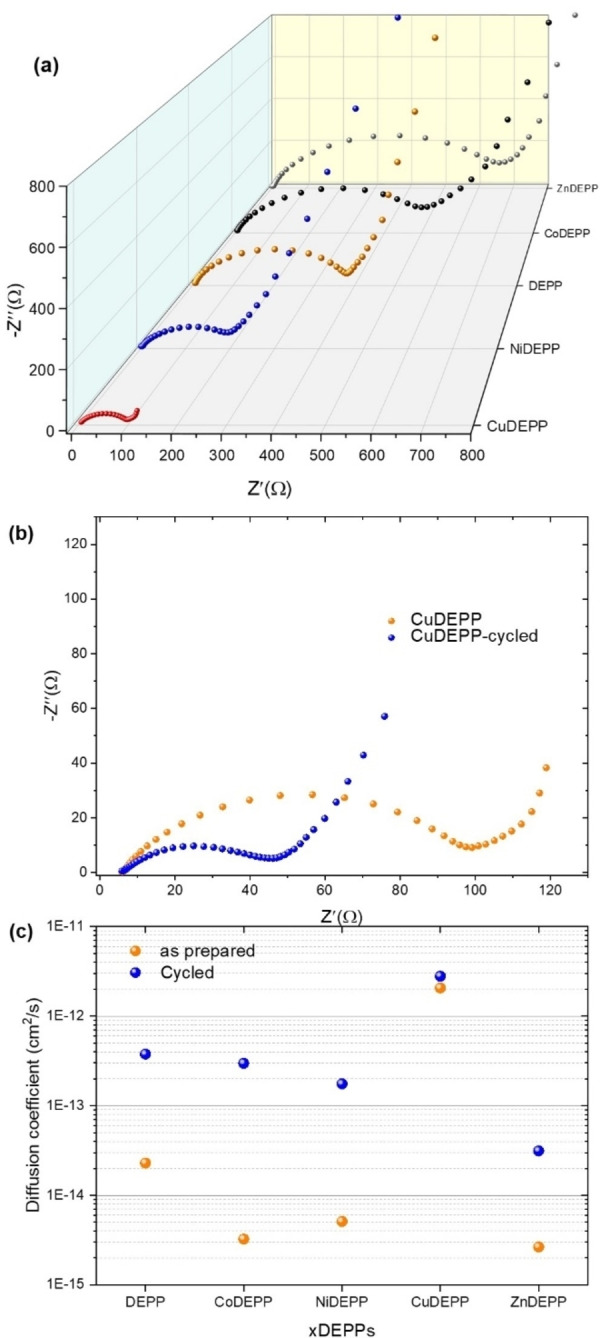
Porphyrin derivatives represent an emerging class of redox-active materials for sustainable electrochemical energy storage. However, their structure-performance relationship is poorly understood, which confines their rational design and thus limits access to their full potential. To gain such understanding, we here focus on the role of the metal ion within porphyrin molecules. The A B -type porphyrin 5,15-bis(ethynyl)-10,20-diphenylporphyrin and its first-row transition metal complexes from Co to Zn are used as models to investigate the relationships between structure and electrochemical performance. It turned out that the choice of central metal atom has a profound influence on the practical voltage window and discharge capacity. The results of DFT calculations suggest that the choice of central metal atom triggers the degree of planarity of the porphyrin. Single crystal diffraction studies illustrate the consequences on the intramolecular rearrangement and packing of metalloporphyrins. Besides the direct effect of the metal choice on the undesired solubility, efficient packing and crystallinity are found to dictate the rate capability and the ion diffusion along with the porosity. Such findings open up a vast space of compositions and morphologies to accelerate the practical application of resource-friendly cathode materials to satisfy the rapidly increasing need for efficient electrical energy storage.
卟啉衍生物是一类新兴的氧化还原活性材料,可用于可持续电化学储能。然而,人们对其结构与性能的关系知之甚少,这限制了其合理设计,从而限制了其充分发挥潜力。为了深入了解这一问题,我们专注于卟啉分子中金属离子的作用。我们选用 A B 型卟啉 5,15-双(乙炔基)-10,20-二苯基卟啉及其从钴到锌的第一过渡系金属配合物作为模型,研究结构与电化学性能之间的关系。结果表明,中心金属原子的选择对实际电压窗口和放电容量有深远影响。密度泛函理论(DFT)计算结果表明,中心金属原子的选择触发了卟啉的平面化程度。单晶衍射研究说明了金属卟啉的分子内重排和堆积的后果。除了金属选择对溶解度的直接影响外,还发现高效堆积和结晶度决定了倍率性能以及离子在多孔材料中的扩散。这些发现为加速资源友好型阴极材料的实际应用开辟了广阔的空间,以满足对高效电能存储日益增长的需求。