Güvenoğlu Merve, Şimşek-Kiper Pelin Özlem, Koşukcu Can, Taskiran Ekim Z, Saltık-Temizel İnci Nur, Gucer Safak, Utine Eda, Boduroğlu Koray
Department of Pediatric Genetics, Hacettepe University Faculty of Medicine, Ankara, Turkey.
Department of Bioinformatics, Institute of Health Sciences, Hacettepe University, Ankara, Turkey.
Pediatr Gastroenterol Hepatol Nutr. 2022 Nov;25(6):441-452. doi: 10.5223/pghn.2022.25.6.441. Epub 2022 Nov 2.
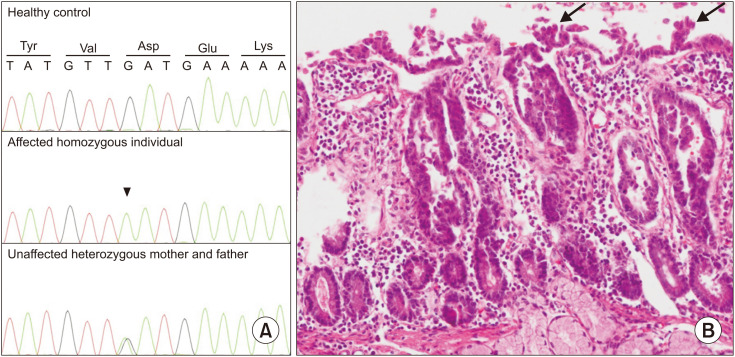
Congenital diarrheal disorders (CDDs) with genetic etiology are uncommon hereditary intestinal diseases characterized by chronic, life-threatening, intractable watery diarrhea that starts in infancy. CDDs can be mechanistically divided into osmotic and secretory diarrhea. Congenital tufting enteropathy (CTE), also known as intestinal epithelial dysplasia, is a type of secretory CDD. CTE is a rare autosomal recessive enteropathy that presents with intractable neonatal-onset diarrhea, intestinal failure, severe malnutrition, and parenteral nutrition dependence. Villous atrophy of the intestinal epithelium, crypt hyperplasia, and irregularity of surface enterocytes are the specific pathological findings of CTE. The small intestine and occasionally the colonic mucosa include focal epithelial tufts. In 2008, Sivagnanam et al. discovered that mutations in the epithelial cell adhesion molecule (, MIM# 185535) were the genetic cause of CTE (MIM# 613217). More than a hundred mutations have been reported to date. Furthermore, mutations in the serine peptidase inhibitor Kunitz type 2 (, MIM# 605124) have been linked to syndromic CTE. In this study, we report the case of a 17-month-old male infant with congenital diarrhea. Despite extensive etiological workup, no etiology could be established before admission to our center. The patient died 15 hours after being admitted to our center in a metabolically decompensated state, probably due to a delay in admission and diagnosis. Molecular autopsy with exome sequencing revealed a previously reported homozygous missense variant, c.757G>A, in , which was confirmed by histopathological examination.
具有遗传病因的先天性腹泻疾病(CDDs)是罕见的遗传性肠道疾病,其特征为始于婴儿期的慢性、危及生命、难治性水样腹泻。CDDs在机制上可分为渗透性腹泻和分泌性腹泻。先天性簇绒性肠病(CTE),也称为肠道上皮发育异常,是分泌性CDD的一种类型。CTE是一种罕见的常染色体隐性肠病,表现为难治性新生儿期腹泻、肠道衰竭、严重营养不良和肠外营养依赖。肠道上皮绒毛萎缩、隐窝增生以及表面肠上皮细胞不规则是CTE的特异性病理表现。小肠以及偶尔结肠黏膜可见局灶性上皮簇。2008年,西瓦格纳南等人发现上皮细胞黏附分子(,MIM编号:185535)的突变是CTE(MIM编号:613217)的遗传病因。迄今为止已报道了一百多种突变。此外,丝氨酸蛋白酶抑制剂库尼茨型2(,MIM编号:605124)的突变与综合征性CTE有关。在本研究中,我们报告了一例17个月大患有先天性腹泻的男婴病例。尽管进行了广泛的病因检查,但在入住我们中心之前仍未明确病因。该患者在以代谢失代偿状态入住我们中心15小时后死亡,可能是由于入院和诊断延迟。通过外显子组测序进行的分子尸检发现了一个先前报道的纯合错义变异,c.757G>A,该变异经组织病理学检查得以证实。