Department of Pediatrics, Division of Nephrology, Boston Children's Hospital, Boston & Harvard Medical School, Boston, MA 02115.
Kidney Disease Initiative, Broad Institute of MIT and Harvard, Cambridge, MA 02142.
Proc Natl Acad Sci U S A. 2022 Dec 20;119(51):e2212810119. doi: 10.1073/pnas.2212810119. Epub 2022 Dec 12.
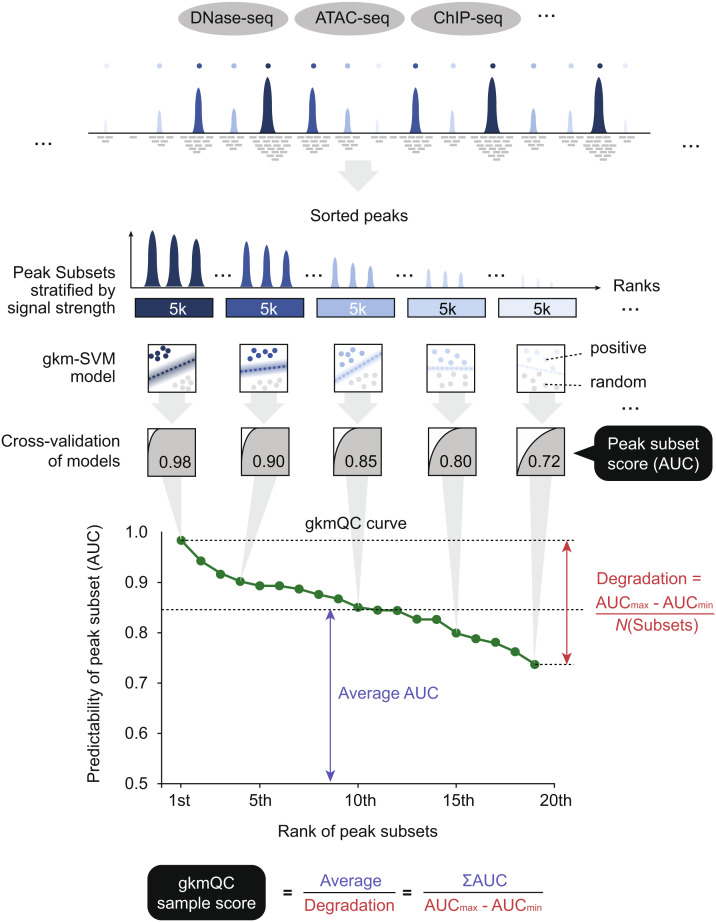
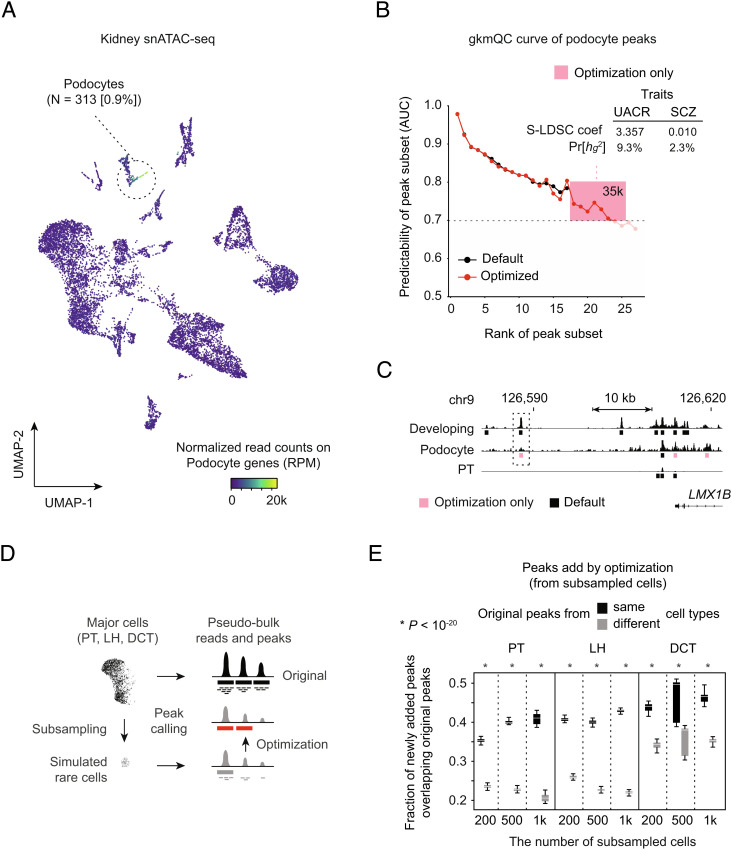
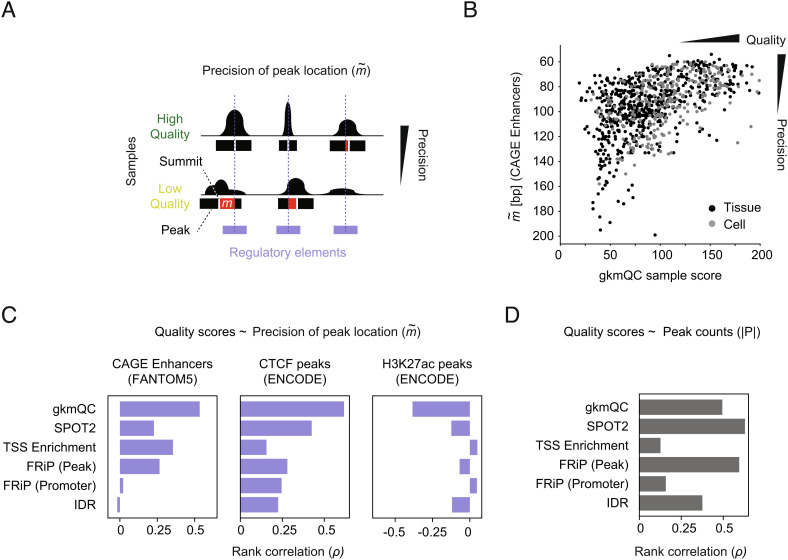
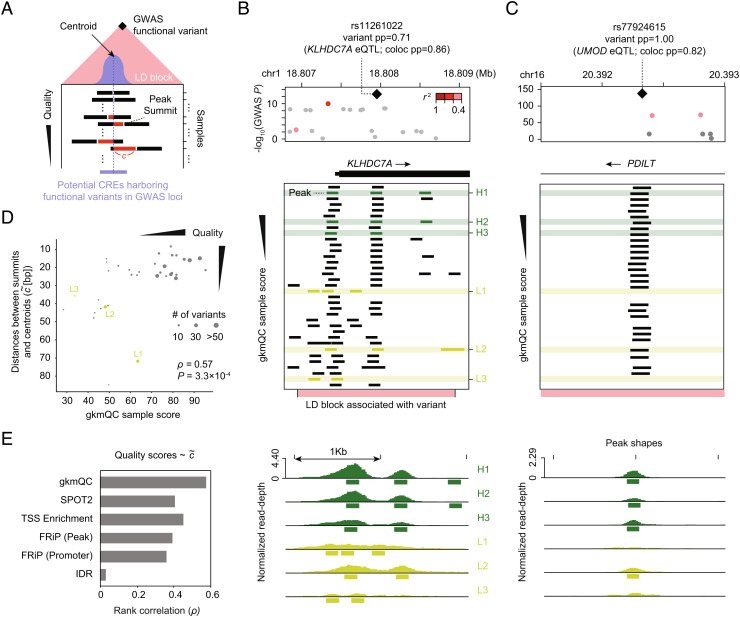
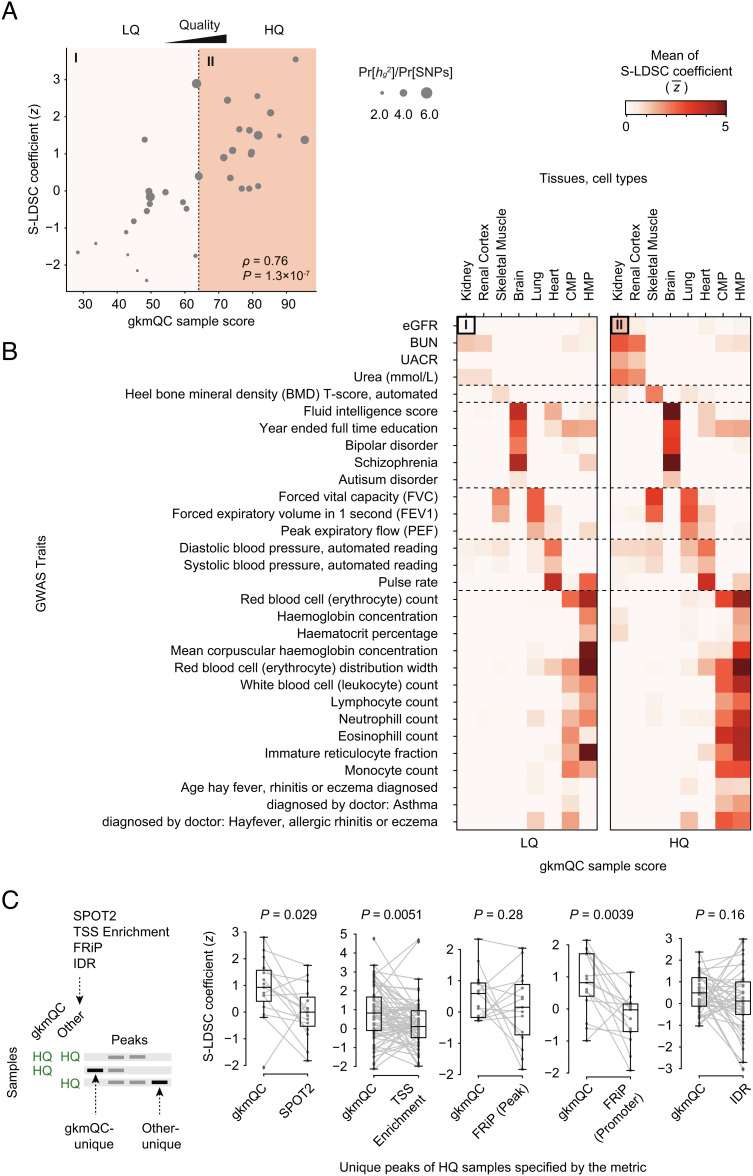
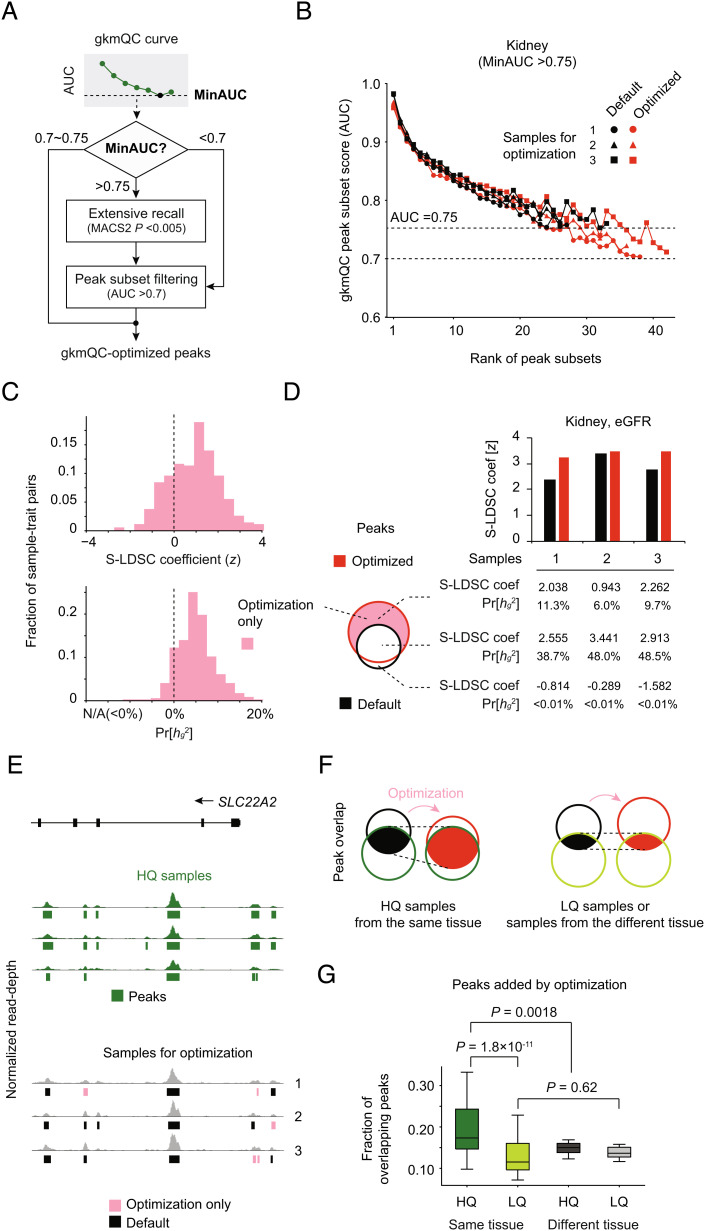
Chromatin accessibility assays are central to the genome-wide identification of gene regulatory elements associated with transcriptional regulation. However, the data have highly variable quality arising from several biological and technical factors. To surmount this problem, we developed a sequence-based machine learning method to evaluate and refine chromatin accessibility data. Our framework, gapped k-mer SVM quality check (gkmQC), provides the quality metrics for a sample based on the prediction accuracy of the trained models. We tested 886 DNase-seq samples from the ENCODE/Roadmap projects to demonstrate that gkmQC can effectively identify "high-quality" (HQ) samples with low conventional quality scores owing to marginal read depths. Peaks identified in HQ samples are more accurately aligned at functional regulatory elements, show greater enrichment of regulatory elements harboring functional variants, and explain greater heritability of phenotypes from their relevant tissues. Moreover, gkmQC can optimize the peak-calling threshold to identify additional peaks, especially for rare cell types in single-cell chromatin accessibility data.
染色质可及性分析是全基因组鉴定与转录调控相关的基因调控元件的核心方法。然而,由于多种生物学和技术因素的影响,数据质量具有高度可变性。为了解决这个问题,我们开发了一种基于序列的机器学习方法来评估和优化染色质可及性数据。我们的框架,缺口 k-mer SVM 质量检查(gkmQC),基于训练模型的预测准确性为样本提供质量指标。我们测试了 ENCODE/Roadmap 项目中的 886 个 DNase-seq 样本,证明 gkmQC 可以有效地识别由于边缘读取深度而导致常规质量分数较低的“高质量”(HQ)样本。在 HQ 样本中鉴定的峰在功能调节元件上的对齐更准确,表现出更多富含具有功能变异的调节元件,并且可以从相关组织中解释更大的表型遗传率。此外,gkmQC 可以优化峰调用阈值来识别更多的峰,特别是在单细胞染色质可及性数据中罕见的细胞类型。