Department of Dermatology, Changsha Hospital of Traditional Chinese Medicine (Changsha Eighth Hospital), Changsha, China.
Department of Dermatology, Hunan Provincial People's Hospital, Changsha, China.
Front Immunol. 2022 Dec 1;13:1054445. doi: 10.3389/fimmu.2022.1054445. eCollection 2022.
A lot of studies have revealed that chronic urticaria (CU) is closely linked with COVID-19. However, there is a lack of further study at the gene level. This research is aimed to investigate the molecular mechanism of COVID-19-related CU bioinformatic ways.
The RNA expression profile datasets of CU (GSE72540) and COVID-19 (GSE164805) were used for the training data and GSE57178 for the verification data. After recognizing the shared differently expressed genes (DEGs) of COVID-19 and CU, genes enrichment, WGCNA, PPI network, and immune infiltration analyses were performed. In addition, machine learning LASSO regression was employed to identify key genes from hub genes. Finally, the networks, gene-TF-miRNA-lncRNA, and drug-gene, of key genes were constructed, and RNA expression analysis was utilized for verification.
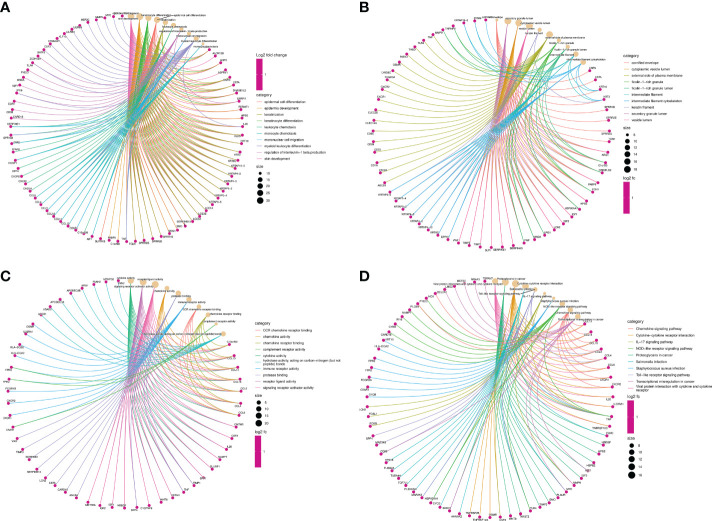
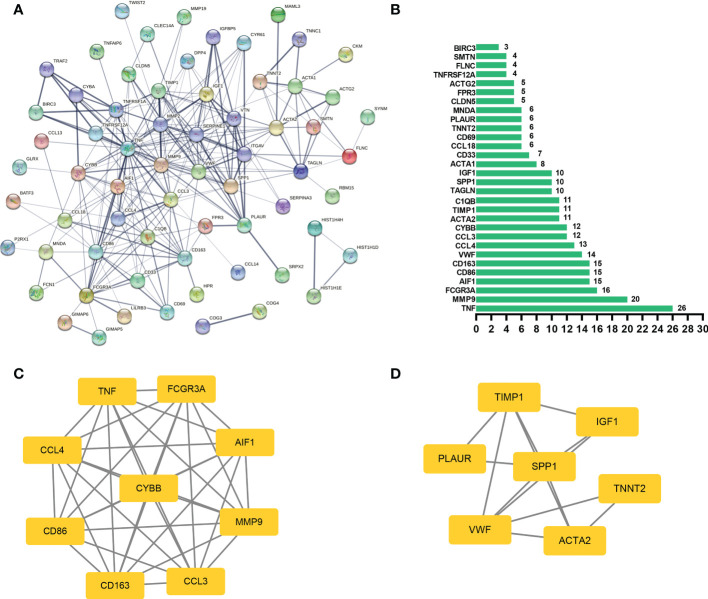
We recognized 322 shared DEGs, and the functional analyses displayed that they mainly participated in immunomodulation of COVID-19-related CU. 9 hub genes (CD86, FCGR3A, AIF1, CD163, CCL4, TNF, CYBB, MMP9, and CCL3) were explored through the WGCNA and PPI network. Moreover, FCGR3A, TNF, and CCL3 were further identified as key genes LASSO regression analysis, and the ROC curves confirmed the dependability of their diagnostic value. Furthermore, our results showed that the key genes were significantly associated with the primary infiltration cells of CU and COVID-19, such as mast cells and macrophages M0. In addition, the key gene-TF-miRNA-lncRNA network was constructed, which contained 46 regulation axes. And most lncRNAs of the network were proved to be a significant expression in CU. Finally, the key gene-drug interaction network, including 84 possible therapeutical medicines, was developed, and their protein-protein docking might make this prediction more feasible.
To sum up, FCGR3A, TNF, and CCL3 might be potential biomarkers for COVID-19-related CU, and the common pathways and related molecules we explored in this study might provide new ideas for further mechanistic research.
大量研究表明,慢性荨麻疹(CU)与 COVID-19 密切相关。然而,在基因水平上的进一步研究还很缺乏。本研究旨在通过生物信息学方法探讨 COVID-19 相关 CU 的分子机制。
采用 CU 的 RNA 表达谱数据集(GSE72540)和 COVID-19 的 RNA 表达谱数据集(GSE164805)作为训练数据,GSE57178 作为验证数据。识别 COVID-19 和 CU 的差异表达基因(DEGs)后,进行基因富集、WGCNA、PPI 网络和免疫浸润分析。此外,采用机器学习 LASSO 回归从 hub 基因中识别关键基因。最后,构建关键基因的网络、基因-TF-miRNA-lncRNA 网络和药物-基因网络,并进行 RNA 表达分析验证。
我们识别出 322 个共享的 DEGs,功能分析表明它们主要参与了 COVID-19 相关 CU 的免疫调节。通过 WGCNA 和 PPI 网络,研究发现了 9 个 hub 基因(CD86、FCGR3A、AIF1、CD163、CCL4、TNF、CYBB、MMP9 和 CCL3)。此外,通过 LASSO 回归分析进一步确定了 FCGR3A、TNF 和 CCL3 为关键基因,ROC 曲线验证了其诊断价值的可靠性。此外,我们的研究结果表明,这些关键基因与 CU 和 COVID-19 的主要浸润细胞,如肥大细胞和巨噬细胞 M0 显著相关。此外,构建了关键基因-TF-miRNA-lncRNA 网络,包含 46 个调节轴。并且,网络中的大多数 lncRNAs 在 CU 中表现出显著的表达。最后,构建了关键基因-药物相互作用网络,包含 84 种可能的治疗药物,其蛋白质-蛋白质对接可能使这一预测更加可行。
综上所述,FCGR3A、TNF 和 CCL3 可能是 COVID-19 相关 CU 的潜在生物标志物,本研究中我们探讨的共同途径和相关分子可能为进一步的机制研究提供新的思路。