Department of Molecular and Translational Medicine, Center of Emphasis in Cancer, Texas Tech University Health Sciences Center El Paso, El Paso, Texas, USA.
L. Frederick Francis Graduate School of Biomedical Sciences, Texas Tech University Health Sciences Center El Paso, El Paso, Texas, USA.
Clin Transl Med. 2022 Dec;12(12):e1146. doi: 10.1002/ctm2.1146.
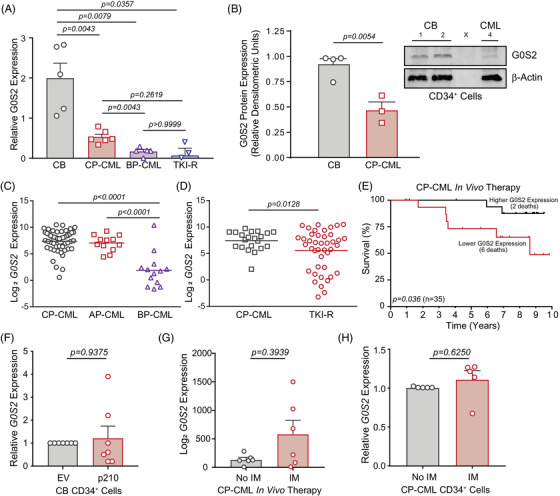
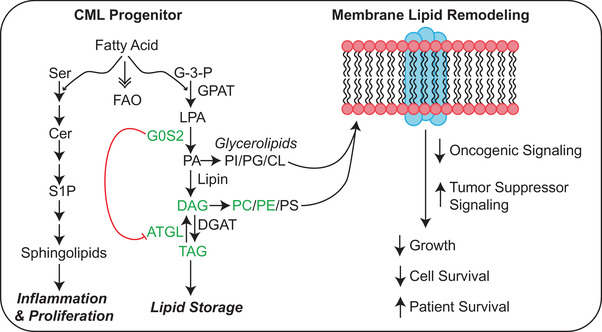
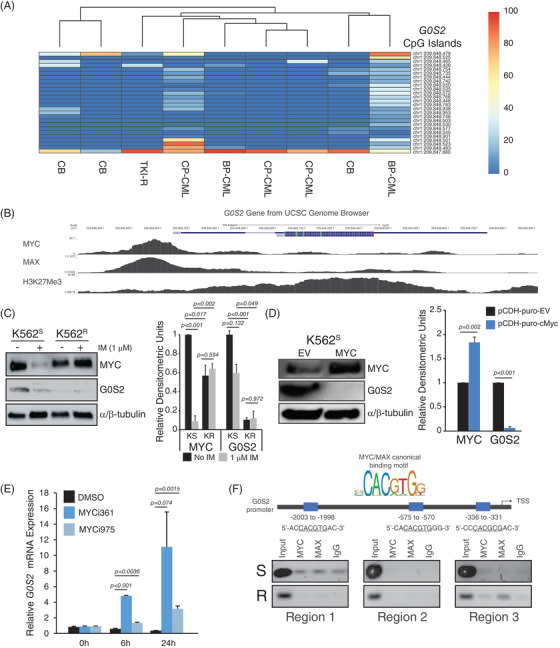
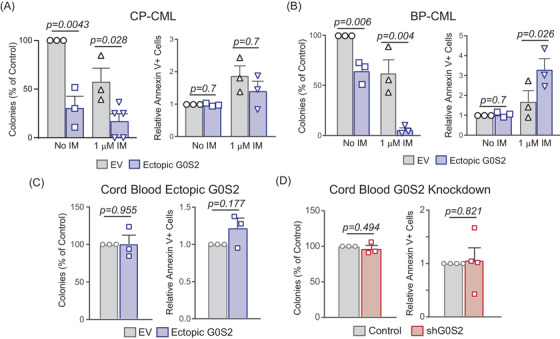
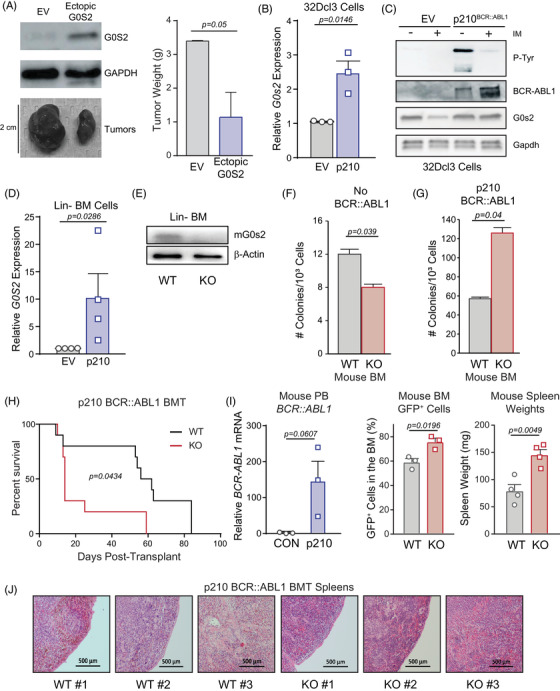
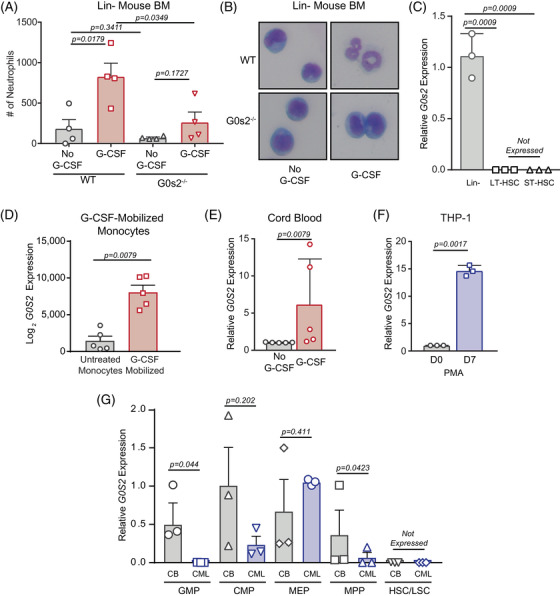
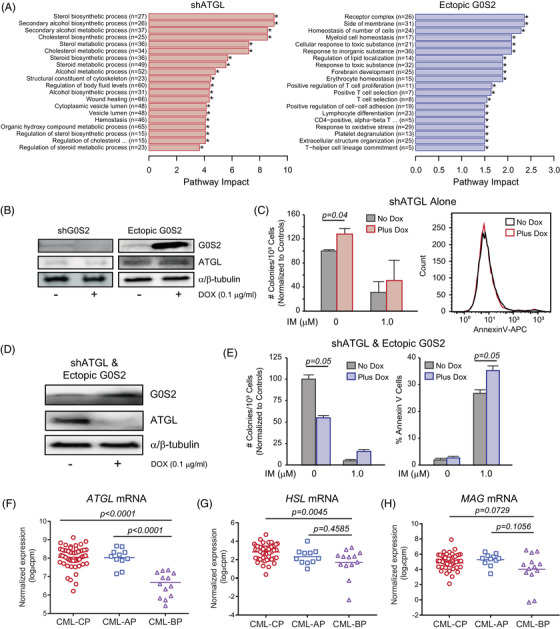
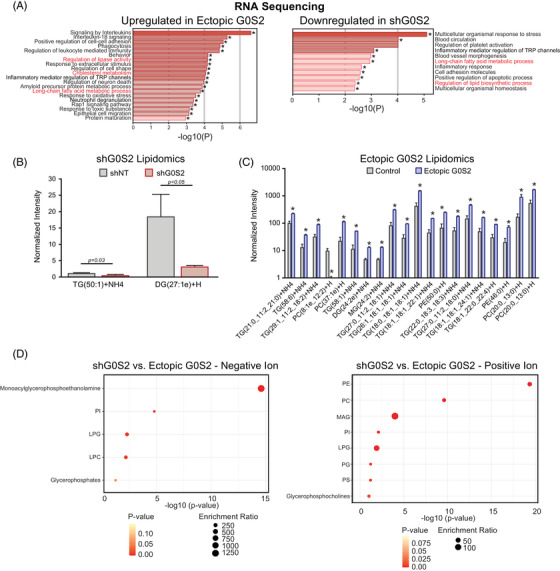
Tyrosine kinase inhibitors (TKIs) targeting BCR::ABL1 have turned chronic myeloid leukaemia (CML) from a fatal disease into a manageable condition for most patients. Despite improved survival, targeting drug-resistant leukaemia stem cells (LSCs) remains a challenge for curative CML therapy. Aberrant lipid metabolism can have a large impact on membrane dynamics, cell survival and therapeutic responses in cancer. While ceramide and sphingolipid levels were previously correlated with TKI response in CML, the role of lipid metabolism in TKI resistance is not well understood. We have identified downregulation of a critical regulator of lipid metabolism, G0/G1 switch gene 2 (G0S2), in multiple scenarios of TKI resistance, including (1) BCR::ABL1 kinase-independent TKI resistance, (2) progression of CML from the chronic to the blast phase of the disease, and (3) in CML versus normal myeloid progenitors. Accordingly, CML patients with low G0S2 expression levels had a worse overall survival. G0S2 downregulation in CML was not a result of promoter hypermethylation or BCR::ABL1 kinase activity, but was rather due to transcriptional repression by MYC. Using CML cell lines, patient samples and G0s2 knockout (G0s2 ) mice, we demonstrate a tumour suppressor role for G0S2 in CML and TKI resistance. Our data suggest that reduced G0S2 protein expression in CML disrupts glycerophospholipid metabolism, correlating with a block of differentiation that renders CML cells resistant to therapy. Altogether, our data unravel a new role for G0S2 in regulating myeloid differentiation and TKI response in CML, and suggest that restoring G0S2 may have clinical utility.
针对 BCR::ABL1 的酪氨酸激酶抑制剂 (TKI) 已将慢性髓系白血病 (CML) 从一种致命疾病转变为大多数患者可控制的疾病。尽管生存得到改善,但针对耐药性白血病干细胞 (LSCs) 的靶向治疗仍然是治愈 CML 治疗的一个挑战。异常的脂质代谢会对癌症中的膜动力学、细胞存活和治疗反应产生重大影响。虽然先前已证实 CML 中神经酰胺和鞘脂水平与 TKI 反应相关,但脂质代谢在 TKI 耐药中的作用尚不清楚。我们已经在多种 TKI 耐药情况下鉴定出脂质代谢关键调节因子 G0/G1 开关基因 2 (G0S2) 的下调,包括 (1) BCR::ABL1 激酶非依赖性 TKI 耐药,(2) CML 从慢性期向疾病爆发期的进展,以及 (3) CML 与正常髓样祖细胞。因此,G0S2 表达水平低的 CML 患者总生存期较差。CML 中 G0S2 的下调不是由于启动子超甲基化或 BCR::ABL1 激酶活性,而是由于 MYC 的转录抑制。使用 CML 细胞系、患者样本和 G0s2 敲除 (G0s2 ) 小鼠,我们证明了 G0S2 在 CML 和 TKI 耐药中的肿瘤抑制作用。我们的数据表明,CML 中 G0S2 蛋白表达减少会破坏甘油磷脂代谢,与阻止分化相关,使 CML 细胞对治疗产生耐药性。总之,我们的数据揭示了 G0S2 在调节髓样分化和 CML 中 TKI 反应中的新作用,并表明恢复 G0S2 可能具有临床应用价值。