Wang Han-Lu, Ruan Dan-Dan, Wu Min, Ji Yuan-Yuan, Hu Xing-Xing, Wu Qiu-Yan, Zhang Yan-Ping, Lin Bin, Hu Ya-Nan, Wang Hang, Tang Yi, Fang Zhu-Ting, Luo Jie-Wei, Liao Li-Sheng, Gao Mei-Zhu
Fujian Provincial Hospital, Shengli Clinical Medical College of Fujian Medical University, Fuzhou, 350001, China.
Department of Cardiovascular Medicine, Fujian Provincial Hospital, Fuzhou, 350001, China.
Thromb J. 2023 Jan 9;21(1):3. doi: 10.1186/s12959-022-00443-6.
Antithrombin (AT) is the main physiological anticoagulant involved in hemostasis. Hereditary AT deficiency is a rare autosomal dominant thrombotic disease mainly caused by mutations in SERPINC1, which was usually manifested as venous thrombosis and pulmonary embolism. In this study, we analyzed the clinical characteristics and screened for mutant genes in two pedigrees with hereditary AT deficiency, and the functional effects of the pathogenic mutations were evaluated.
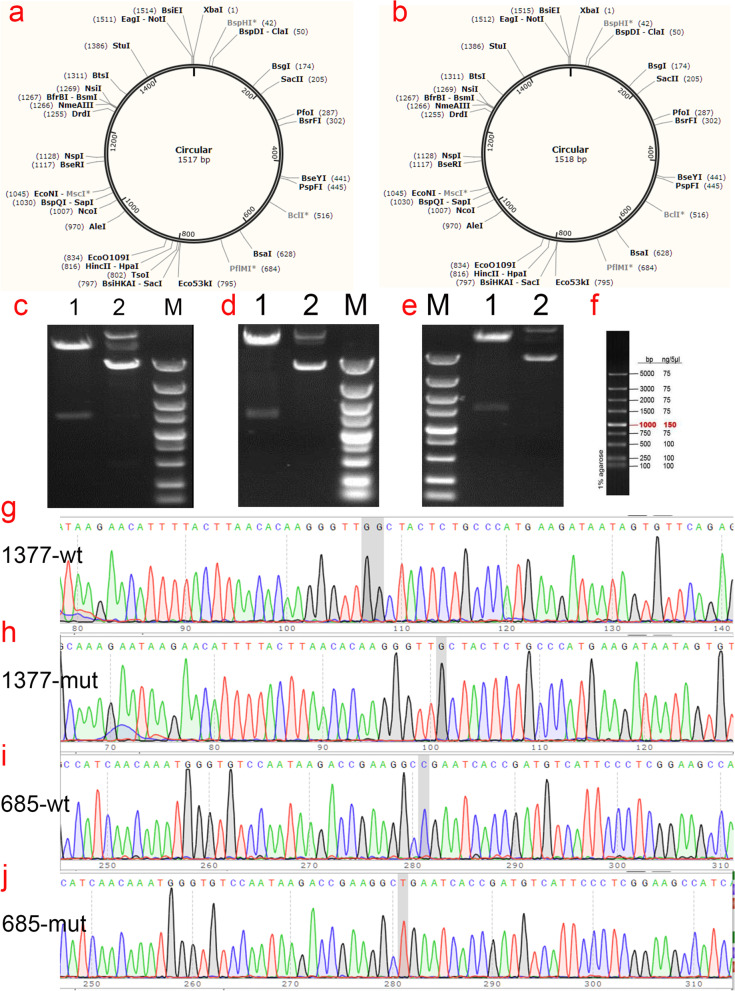
Candidate gene variants were analyzed by next-generation sequencing to screen pathogenic mutations in probands, followed by segregation analysis in families by Sanger sequencing. Mutant and wild-type plasmids were constructed and transfected into HEK293T cells to observe protein expression and cellular localization of SERPINC1. The structure and function of the mutations were analyzed by bioinformatic analyses.
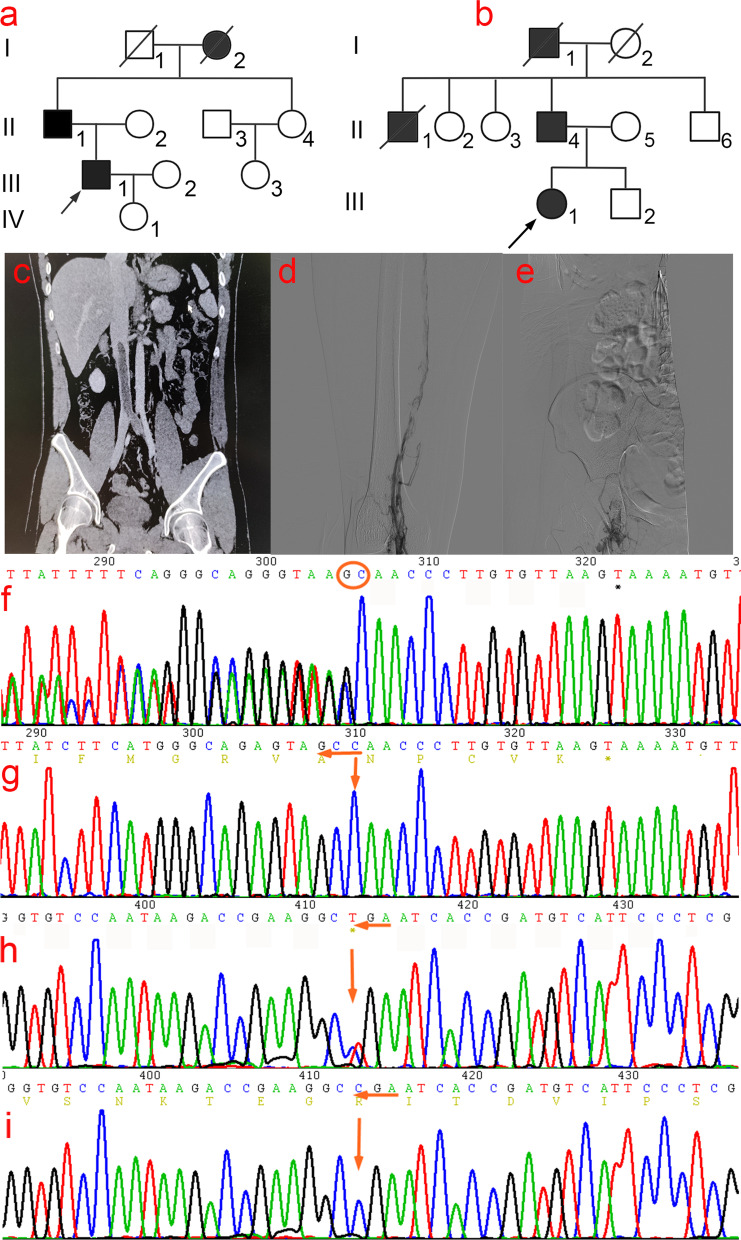
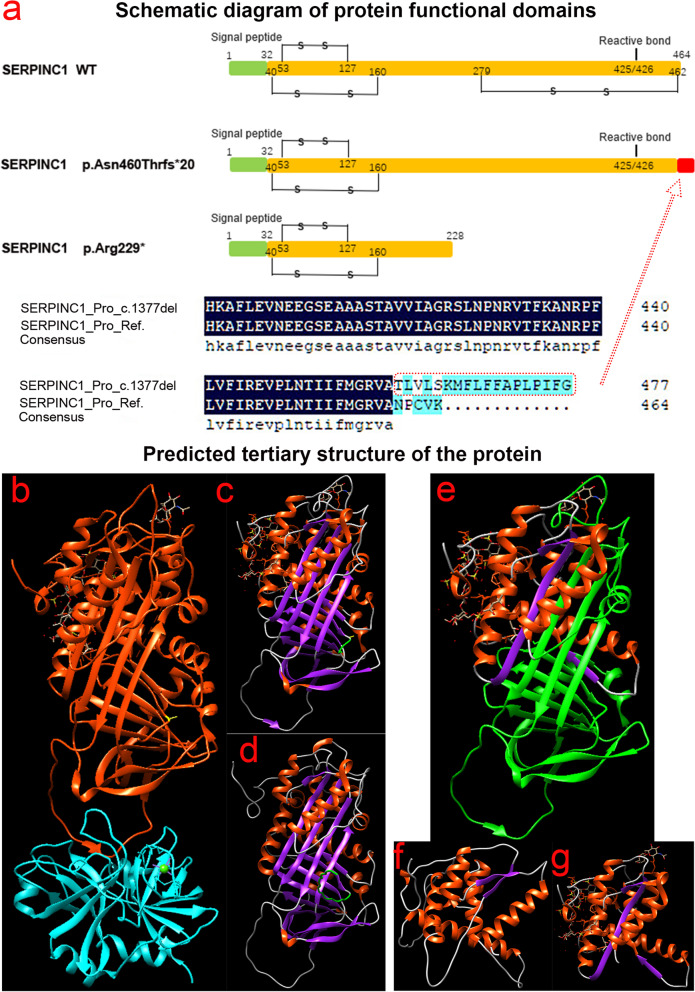
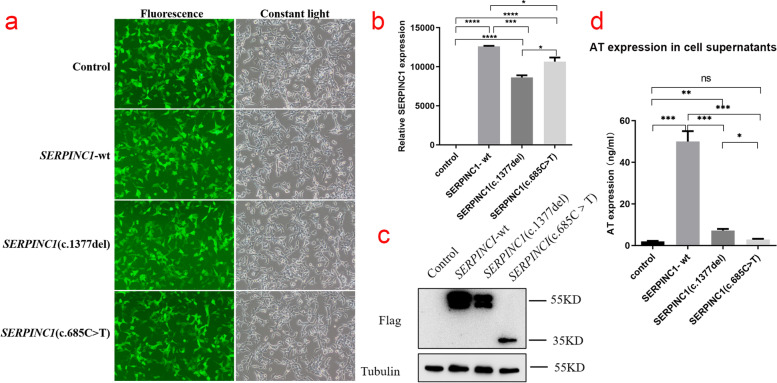
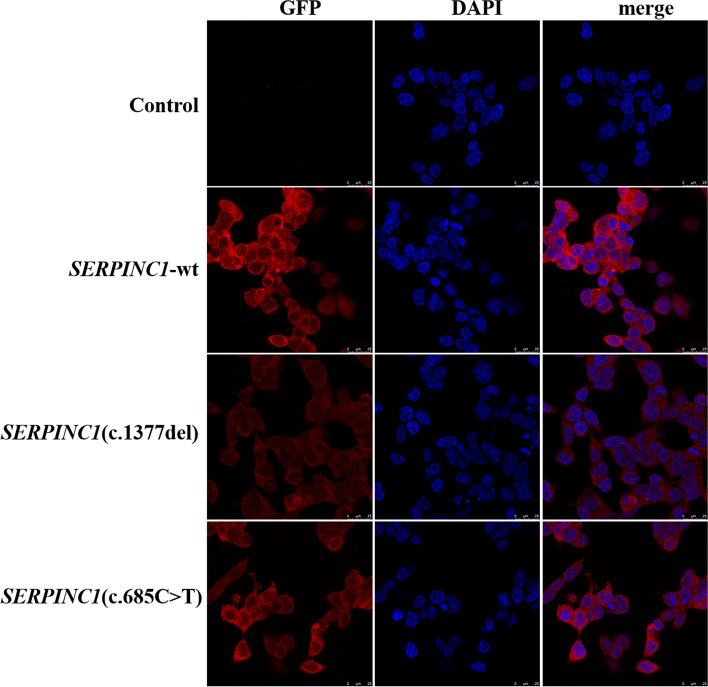
The proband of pedigree A with AT deficiency carried a heterozygous frameshift mutation c.1377delC (p.Asn460Thrfs20) in SERPINC1 (NM000488.3), a 1377C base deletion in exon 7 resulting in a backward shift of the open reading frame, with termination after translation of 20 residues, and a different residue sequence translated after the frameshift. Bioinformatics analysis suggests that the missing amino acid sequence caused by the frameshift mutation might disrupt the disulfide bond between Cys279 and Cys462 and affect the structural function of the protein. This newly discovered variant is not currently included in the ClinVar and HGMD databases. p.Arg229 resulted in a premature stop codon in exon 4, and bioinformatics analysis suggests that the truncated protein structure lost its domain of interaction with factor IX (Ala414 site) after the deletion of nonsense mutations. However, considering the AT truncation protein resulting from the p.Arg229* variant loss a great proportion of the molecule, we speculate the variant may affect two functional domains HBS and RCL and lack of the corresponding function. The thrombophilia and decreased-AT-activity phenotypes of the two pedigrees were separated from their genetic variants. After lentiviral plasmid transfection into HEK293T cells, the expression level of AT protein decreased in the constructed c.1377delC mutant cells compared to that in the wild-type, which was not only reduced in c.685C > T mutant cells but also showed a significant band at 35 kDa, suggesting a truncated protein. Immunofluorescence localization showed no significant differences in protein localization before and after the mutation.
The p.Asn460Thrfs20 and p.Arg229 variants of SERPINC1 were responsible for the two hereditary AT deficiency pedigrees, which led to AT deficiency by different mechanisms. The p.Asn460Thrfs*20 variant is reported for the first time.
抗凝血酶(AT)是参与止血的主要生理性抗凝剂。遗传性AT缺乏症是一种罕见的常染色体显性血栓形成疾病,主要由SERPINC1基因突变引起,通常表现为静脉血栓形成和肺栓塞。在本研究中,我们分析了两个遗传性AT缺乏症家系的临床特征并筛选突变基因,评估了致病突变的功能影响。
通过下一代测序分析候选基因变异,以筛选先证者中的致病突变,随后通过桑格测序在家族中进行分离分析。构建突变型和野生型质粒并转染至HEK293T细胞中,观察SERPINC1的蛋白表达和细胞定位。通过生物信息学分析对突变的结构和功能进行分析。
家系A中患有AT缺乏症的先证者在SERPINC1(NM000488.3)中携带杂合移码突变c.1377delC(p.Asn460Thrfs20),外显子7中的1377C碱基缺失导致开放阅读框向后移位,翻译20个残基后终止,移码后翻译出不同的残基序列。生物信息学分析表明,移码突变导致的缺失氨基酸序列可能破坏Cys279和Cys462之间的二硫键,并影响蛋白质的结构功能。这个新发现的变异目前未包含在ClinVar和HGMD数据库中。p.Arg229导致外显子4中出现提前终止密码子,生物信息学分析表明,截短的蛋白质结构在缺失无义突变后失去了与因子IX(Ala414位点)相互作用的结构域。然而,考虑到由p.Arg229*变异产生的AT截短蛋白失去了很大一部分分子,我们推测该变异可能影响两个功能结构域HBS和RCL并缺乏相应功能。两个家系的血栓形成倾向和AT活性降低的表型与其遗传变异分离。慢病毒质粒转染至HEK293T细胞后,与野生型相比,构建的c.1377delC突变细胞中AT蛋白表达水平降低,c.685C>T突变细胞中不仅降低,还在35 kDa处出现明显条带,提示存在截短蛋白。免疫荧光定位显示突变前后蛋白质定位无显著差异。
SERPINC1的p.Asn460Thrfs20和p.Arg229变异导致了两个遗传性AT缺乏症家系,通过不同机制导致AT缺乏。p.Asn460Thrfs*20变异为首次报道。