MRC Human Immunology Unit, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
Department of Immunobiology, King's College London, London, United Kingdom.
J Immunol. 2023 Mar 1;210(5):547-557. doi: 10.4049/jimmunol.2200212.
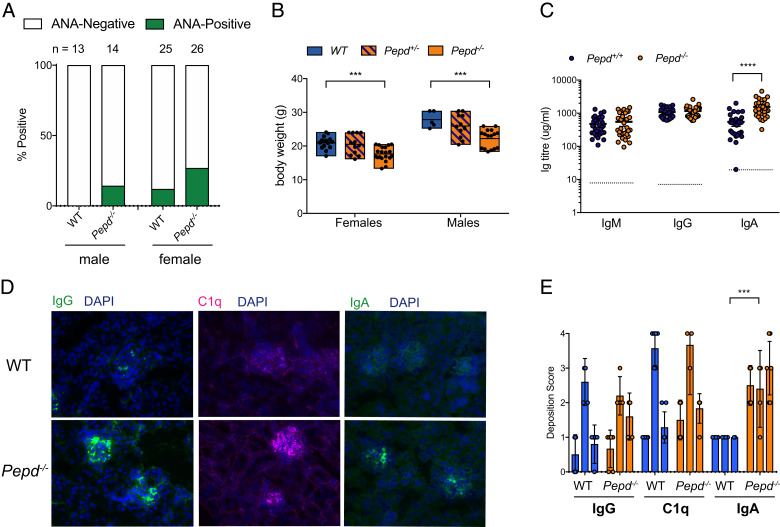
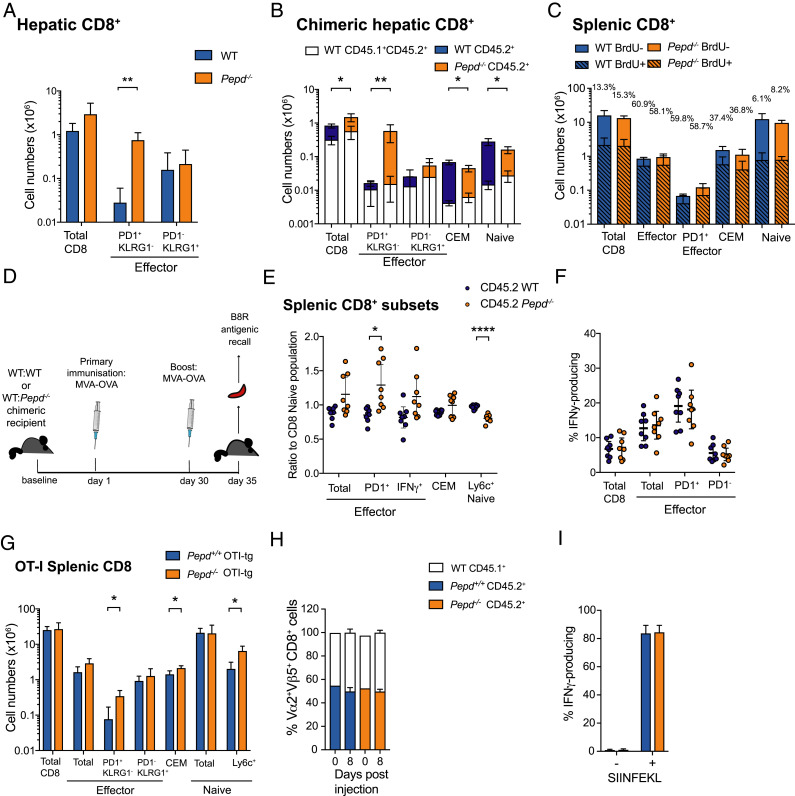
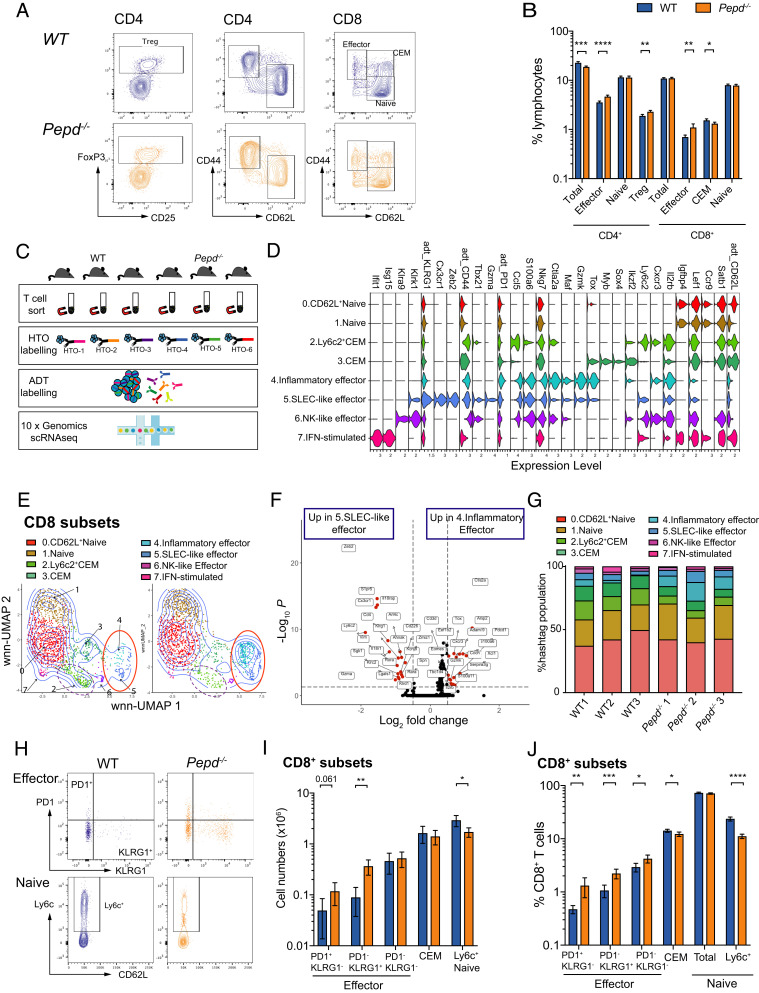
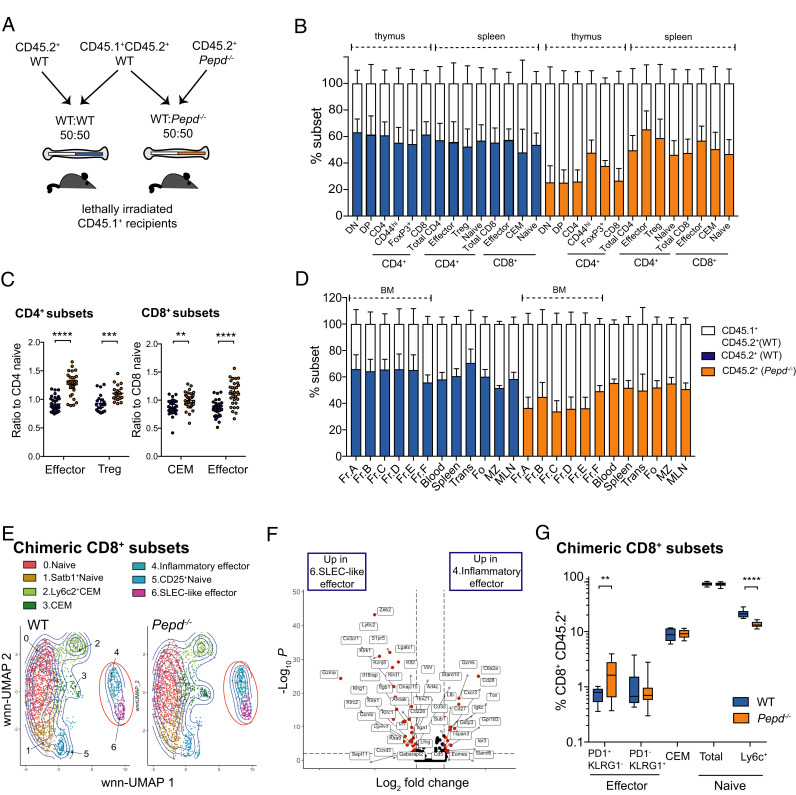
Prolidase deficiency (PD) is a multisystem disorder caused by mutations in the PEPD gene, which encodes a ubiquitously expressed metallopeptidase essential for the hydrolysis of dipeptides containing C-terminal proline or hydroxyproline. PD typically presents in childhood with developmental delay, skin ulcers, recurrent infections, and, in some patients, autoimmune features that can mimic systemic lupus erythematosus. The basis for the autoimmune association is uncertain, but might be due to self-antigen exposure with tissue damage, or indirectly driven by chronic infection and microbial burden. In this study, we address the question of causation and show that Pepd-null mice have increased antinuclear autoantibodies and raised serum IgA, accompanied by kidney immune complex deposition, consistent with a systemic lupus erythematosus-like disease. These features are associated with an accumulation of CD4 and CD8 effector T cells in the spleen and liver. Pepd deficiency leads to spontaneous T cell activation and proliferation into the effector subset, which is cell intrinsic and independent of Ag receptor specificity or antigenic stimulation. However, an increase in KLRG1+ effector CD8 cells is not observed in mixed chimeras, in which the autoimmune phenotype is also absent. Our findings link autoimmune susceptibility in PD to spontaneous T cell dysfunction, likely to be acting in combination with immune activators that lie outside the hemopoietic system but result from the abnormal metabolism or loss of nonenzymatic prolidase function. This knowledge provides insight into the role of prolidase in the maintenance of self-tolerance and highlights the importance of treatment to control T cell activation.
脯肽酶缺乏症(PD)是一种多系统疾病,由 PEPD 基因的突变引起,该基因编码一种广泛表达的金属肽酶,对于含有 C 末端脯氨酸或羟脯氨酸的二肽的水解至关重要。PD 通常在儿童期表现为发育迟缓、皮肤溃疡、反复感染,并且在一些患者中具有自身免疫特征,可模仿系统性红斑狼疮。自身免疫关联的基础尚不确定,但可能是由于自身抗原暴露伴组织损伤,或者间接由慢性感染和微生物负担驱动。在这项研究中,我们解决了因果关系的问题,并表明 Pepd 基因敲除小鼠具有增加的核抗自身抗体和升高的血清 IgA,伴有肾脏免疫复合物沉积,符合系统性红斑狼疮样疾病。这些特征与脾脏和肝脏中 CD4 和 CD8 效应 T 细胞的积累有关。Pepd 缺乏导致自发的 T 细胞激活和增殖为效应亚群,这是细胞内在的,与 Ag 受体特异性或抗原刺激无关。然而,在混合嵌合体中未观察到 KLRG1+效应 CD8 细胞的增加,其中自身免疫表型也不存在。我们的发现将 PD 中的自身免疫易感性与自发的 T 细胞功能障碍联系起来,可能与免疫激活剂共同作用,这些免疫激活剂位于造血系统之外,但源于异常代谢或非酶促脯肽酶功能的丧失。这一知识为我们了解脯肽酶在维持自身耐受中的作用提供了线索,并强调了治疗以控制 T 细胞激活的重要性。