Department of Biostatistics, Harvard TH Chan School of Public Health, Boston, MA, USA.
Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA, USA.
Sci Rep. 2023 Jan 21;13(1):1197. doi: 10.1038/s41598-022-26434-1.
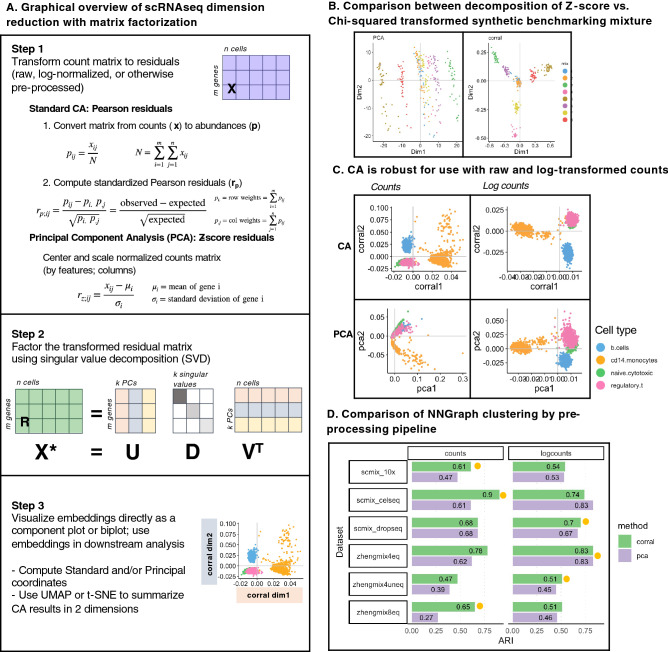
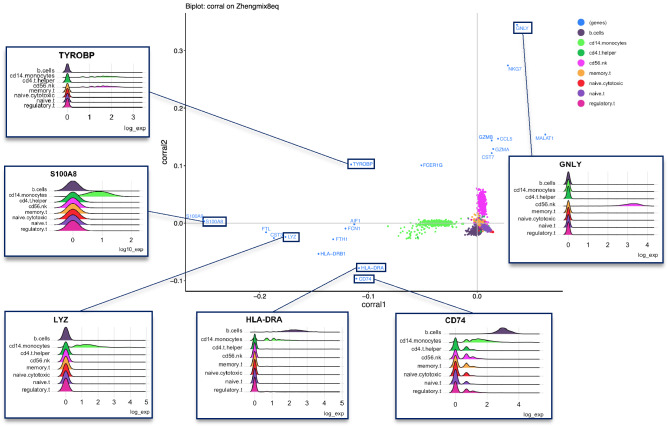
Effective dimension reduction is essential for single cell RNA-seq (scRNAseq) analysis. Principal component analysis (PCA) is widely used, but requires continuous, normally-distributed data; therefore, it is often coupled with log-transformation in scRNAseq applications, which can distort the data and obscure meaningful variation. We describe correspondence analysis (CA), a count-based alternative to PCA. CA is based on decomposition of a chi-squared residual matrix, avoiding distortive log-transformation. To address overdispersion and high sparsity in scRNAseq data, we propose five adaptations of CA, which are fast, scalable, and outperform standard CA and glmPCA, to compute cell embeddings with more performant or comparable clustering accuracy in 8 out of 9 datasets. In particular, we find that CA with Freeman-Tukey residuals performs especially well across diverse datasets. Other advantages of the CA framework include visualization of associations between genes and cell populations in a "CA biplot," and extension to multi-table analysis; we introduce corralm for integrative multi-table dimension reduction of scRNAseq data. We implement CA for scRNAseq data in corral, an R/Bioconductor package which interfaces directly with single cell classes in Bioconductor. Switching from PCA to CA is achieved through a simple pipeline substitution and improves dimension reduction of scRNAseq datasets.
有效的降维对于单细胞 RNA 测序 (scRNAseq) 分析至关重要。主成分分析 (PCA) 被广泛应用,但它需要连续的、正态分布的数据;因此,在 scRNAseq 应用中,它通常与对数转换相结合,这可能会扭曲数据并掩盖有意义的变化。我们描述了对应分析 (CA),这是一种基于计数的 PCA 替代方法。CA 基于卡方残差矩阵的分解,避免了有失真的对数转换。为了解决 scRNAseq 数据中的过分散和高稀疏性问题,我们提出了 CA 的五种适应性方法,这些方法在 9 个数据集的 8 个数据集中,计算细胞嵌入的速度更快、可扩展性更强,并且性能优于标准 CA 和 glmPCA,聚类准确性相当或更高。特别是,我们发现,使用 Freeman-Tukey 残差的 CA 在各种数据集上表现尤其出色。CA 框架的其他优点包括在“CA 双标图”中可视化基因和细胞群体之间的关联,以及扩展到多表分析;我们引入了 corralm,用于整合 scRNAseq 数据的多表降维。我们在 corral 中实现了 CA 用于 scRNAseq 数据,corral 是一个 R/Bioconductor 包,它可以直接与 Bioconductor 中的单细胞类接口。通过简单的流水线替换从 PCA 切换到 CA 可以提高 scRNAseq 数据集的降维效果。