Vogler Emily C, Mahavongtrakul Matthew, Sarkan Kristianna, Bohannan Ryan C, Catuara-Solarz Silvina, Busciglio Jorge
Department of Neurobiology and Behavior, University of California, Irvine, Irvine, CA, United States.
Institute for Memory Impairments and Neurological Disorders, University of California, Irvine, Irvine, CA, United States.
Front Neurol. 2023 Jan 20;13:882635. doi: 10.3389/fneur.2022.882635. eCollection 2022.
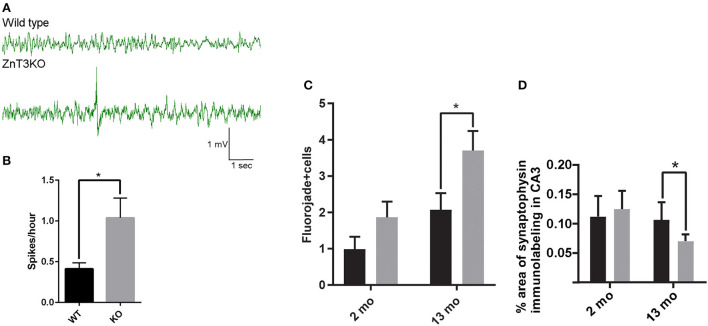
Vesicular Zn (zinc) is released at synapses and has been demonstrated to modulate neuronal responses. However, mechanisms through which dysregulation of zinc homeostasis may potentiate neuronal dysfunction and neurodegeneration are not well-understood. We previously reported that accumulation of soluble amyloid beta oligomers (AβO) at synapses correlates with synaptic loss and that AβO localization at synapses is regulated by synaptic activity and enhanced by the release of vesicular Zn in the hippocampus, a brain region that deteriorates early in Alzheimer's disease (AD). Significantly, drugs regulating zinc homeostasis inhibit AβO accumulation and improve cognition in mouse models of AD. We used both sexes of a transgenic mouse model lacking synaptic Zn (ZnT3KO) that develops AD-like cognitive impairment and neurodegeneration to study the effects of disruption of Zn modulation of neurotransmission in cognition, protein expression and activation, and neuronal excitability. Here we report that the genetic removal of synaptic Zn results in progressive impairment of hippocampal-dependent memory, reduces activity-dependent increase in Erk phosphorylation and BDNF mRNA, alters regulation of Erk activation by NMDAR subunits, increases neuronal spiking, and induces biochemical and morphological alterations consistent with increasing epileptiform activity and neurodegeneration as ZnT3KO mice age. Our study shows that disruption of synaptic Zn triggers neurodegenerative processes and is a potential pathway through which AβO trigger altered expression of neurotrophic proteins, along with reduced hippocampal synaptic density and degenerating neurons, neuronal spiking activity, and cognitive impairment and supports efforts to develop therapeutics to preserve synaptic zinc homeostasis in the brain as potential treatments for AD.
囊泡锌在突触处释放,并且已被证明可调节神经元反应。然而,锌稳态失调可能增强神经元功能障碍和神经退行性变的机制尚未完全明确。我们之前报道过,可溶性淀粉样β寡聚体(AβO)在突触处的积累与突触丧失相关,并且AβO在突触处的定位受突触活动调节,在海马体中囊泡锌的释放会增强这种定位,海马体是在阿尔茨海默病(AD)早期就会发生退化的脑区。重要的是,调节锌稳态的药物可抑制AβO积累并改善AD小鼠模型的认知能力。我们使用了缺乏突触锌的转基因小鼠模型(ZnT3KO)的雌雄小鼠,该模型会出现类似AD的认知障碍和神经退行性变,以研究锌对神经传递的调节被破坏在认知、蛋白质表达与激活以及神经元兴奋性方面的影响。在此我们报告,突触锌的基因去除导致海马依赖性记忆逐渐受损,减少了依赖活动的Erk磷酸化和BDNF mRNA的增加,改变了NMDAR亚基对Erk激活的调节,增加了神经元放电,并随着ZnT3KO小鼠年龄增长诱导了与癫痫样活动增加和神经退行性变一致的生化和形态学改变。我们的研究表明,突触锌的破坏触发了神经退行性过程,并且是AβO触发神经营养蛋白表达改变的潜在途径,同时伴有海马突触密度降低和神经元退化、神经元放电活动以及认知障碍,并支持开发治疗方法以维持大脑中突触锌稳态作为AD潜在治疗手段的努力。