Xu Yang, Kramann Rafael, McCord Rachel Patton, Hayat Sikander
UT-ORNL Graduate School of Genome Science and Technology, University of Tennessee, Knoxville, TN 37996, USA.
Institute of Experimental Medicine and Systems Biology, RWTH Aachen University, Aachen, Germany.
Res Sq. 2023 Jan 23:rs.3.rs-2485985. doi: 10.21203/rs.3.rs-2485985/v1.
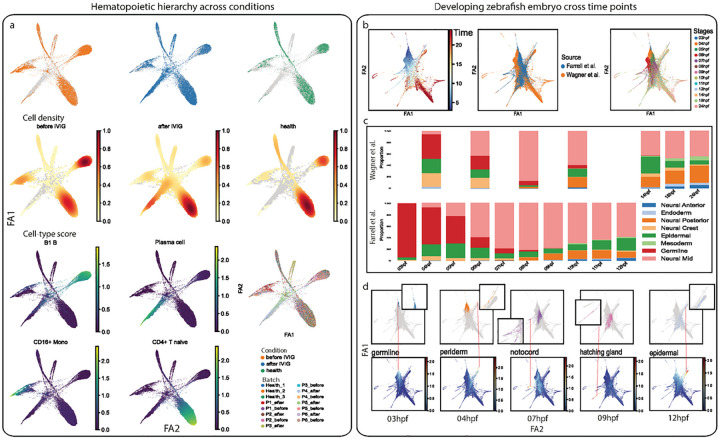
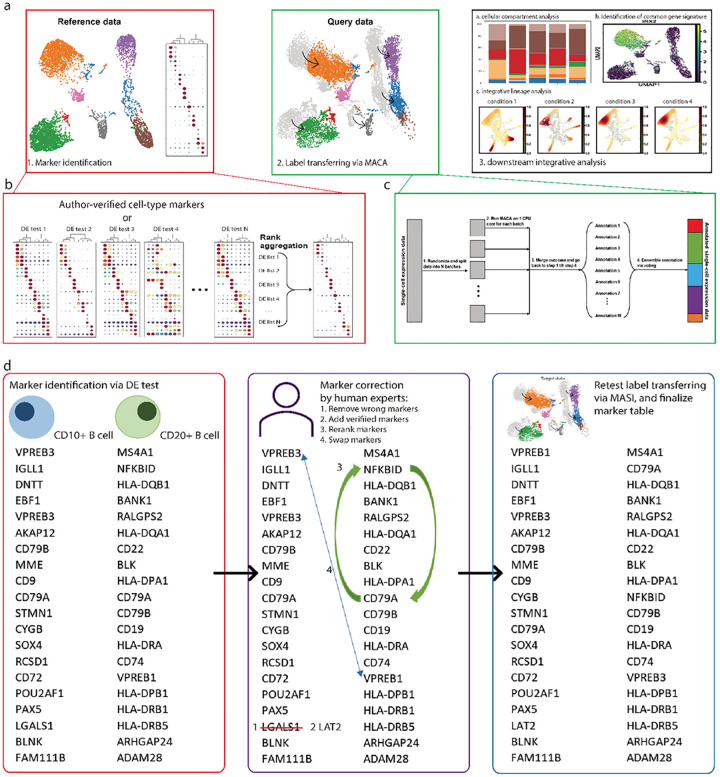
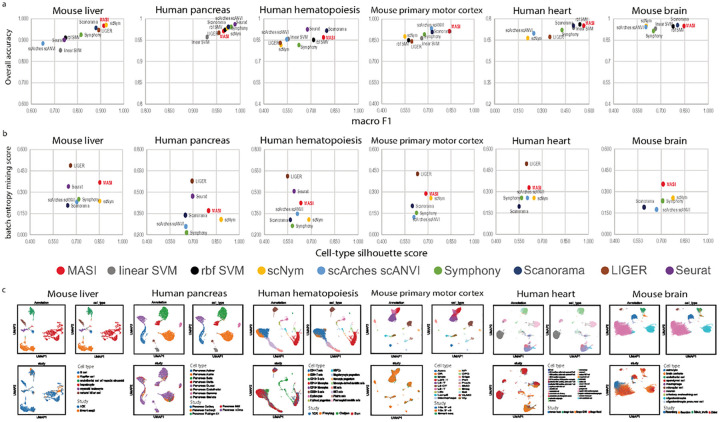
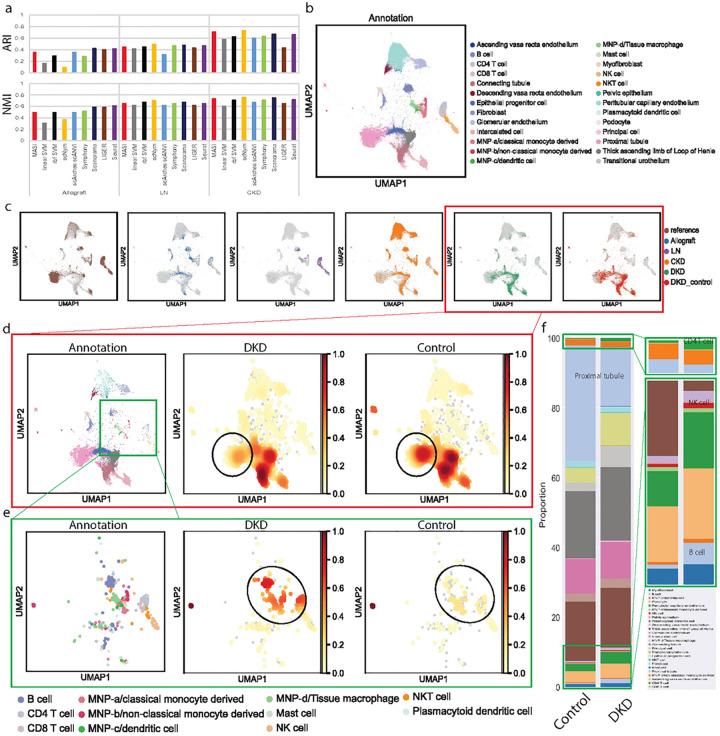
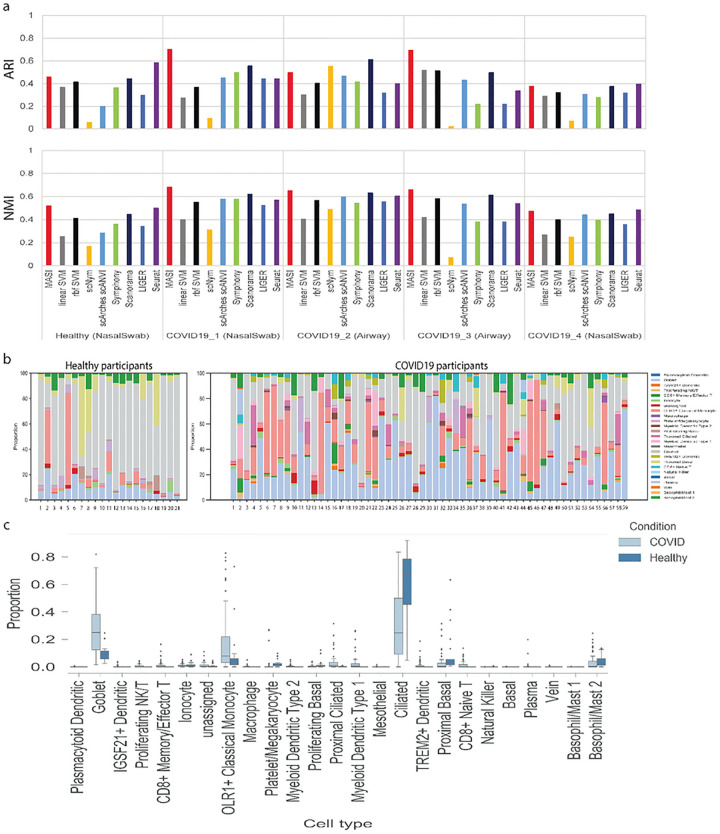
Single-cell transcriptomics datasets from the same anatomical sites generated by different research labs are becoming increasingly common. However, fast and computationally inexpensive tools for standardization of cell-type annotation and data integration are still needed in order to increase research inclusivity. To standardize cell-type annotation and integrate single-cell transcriptomics datasets, we have built a fast model-free integration method, named MASI (Marker-Assisted Standardization and Integration). MASI first identifies putative cell-type markers from reference data through an ensemble approach. Then, it converts gene expression matrix to cell-type score matrix with the identified putative cell-type markers for the purpose of cell-type annotation and data integration. Because of integration through cell-type markers instead of model inference, MASI can annotate approximately one million cells on a personal laptop, which provides a cheap computational alternative for the single-cell community. We benchmark MASI with other well-established methods and demonstrate that MASI outperforms other methods based on speed. Its performance for both tasks of data integration and cell-type annotation are comparable or even superior to these existing methods. To harness knowledge from single-cell atlases, we demonstrate three case studies that cover integration across biological conditions, surveyed participants, and research groups, respectively.
不同研究实验室生成的来自相同解剖部位的单细胞转录组学数据集正变得越来越普遍。然而,为了提高研究的包容性,仍然需要快速且计算成本低廉的工具来实现细胞类型注释的标准化和数据整合。为了标准化细胞类型注释并整合单细胞转录组学数据集,我们构建了一种快速的无模型整合方法,名为MASI(标记辅助标准化和整合)。MASI首先通过一种集成方法从参考数据中识别推定的细胞类型标记。然后,为了进行细胞类型注释和数据整合,它使用所识别的推定细胞类型标记将基因表达矩阵转换为细胞类型评分矩阵。由于是通过细胞类型标记而非模型推理进行整合,MASI可以在个人笔记本电脑上注释大约一百万个细胞,这为单细胞研究群体提供了一种成本低廉的计算替代方案。我们用其他成熟的方法对MASI进行了基准测试,并证明MASI在速度方面优于其他方法。它在数据整合和细胞类型注释这两项任务上的性能与现有方法相当,甚至更优。为了利用单细胞图谱中的知识,我们展示了三个案例研究,分别涵盖了跨生物条件、被调查参与者和研究组的整合。