Ibrahim Muktar Musa, Uzairu Adamu, Ibrahim Muhammad Tukur, Umar Abdullahi Bello
Department of Chemistry, Faculty of Physical Sciences, Ahmadu Bello University P. M. B 1045 Zaria Nigeria
RSC Adv. 2023 Jan 24;13(6):3402-3415. doi: 10.1039/d2ra07382j.
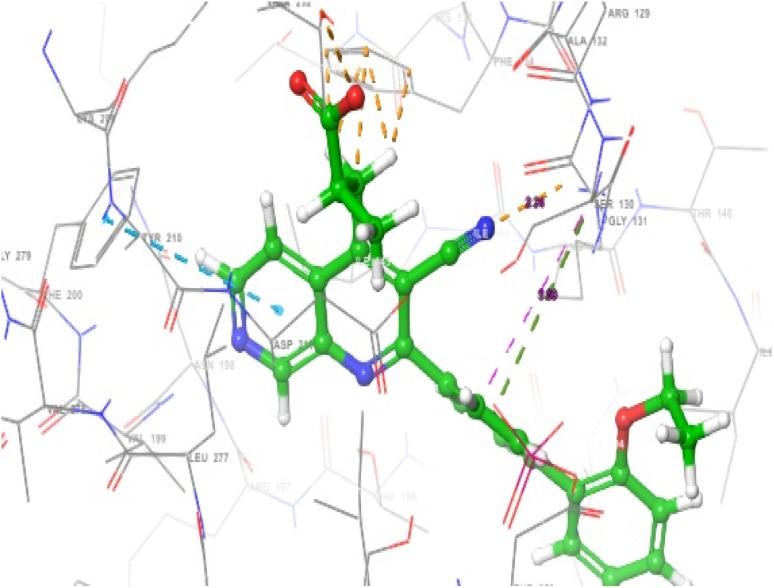
PIP4K2A is a type II lipid kinase that catalyzed the rate-limiting step of the conversion of phosphatidylinositol-5-phosphate (PI5P) into phosphatidylinositol 4,5-bisphosphate (PI4,5P2). PIP4K2A has been intricately linked to the inhibition of various types of tumors reactive oxygen species-mediated apoptosis, making it an important therapeutic target. In the quest of finding biologically active substances with efficient PIP4K2A inhibitory activity, machine learning algorithms were used to investigate the quantitative relationship between structures and inhibitory activities of 1,7-naphthyridine analogues. Three machine learning algorithms (MLR, ANN, and SVM) were used to develop QSAR models that can effectively predict the PIP4K2A inhibitory activity of a library of 1,7-naphthyridine analogues. The cascaded feature selection method was performed by sequential application of GFA and MP5 algorithms to identify a molecular descriptor subset that can best describe the PIP4K2A inhibitory activity of 1,7-naphthyridine analogues. PIP4K2A inhibitory activities predicted by the ML models were strongly correlated with the experimental values. The QSAR Modelling indicates that the best-performing ML model was SVM with the RBF kernel function. The SVM model performed very well in predicting PIP4K2A inhibitory activity of the 1,7-naphthyridine analogues with RTR and QEX values of 0.9845 and 0.8793 respectively. To further gain more structural insight into the origin of PIP4K2A inhibitory activity of 1,7-naphthyridine analogues, molecular docking studies were performed. The results indicate that five compounds; 15, 25, 13, 09, and 28 were found to have a high binding affinity with the receptor molecules. Hydrogen bonding, pi-pi interaction, and pi-cation interactions were found to modulate the binding interaction of the inhibitors. Although the SVM gives essentially a black-box model which cannot be readily interpreted, using SVM in tandem with MLR and ANN provides a unique perspective in building robust QSAR predictive models. The superior predictive performance of the ML models and the explanatory power of MLR models were combined to provide a unique insight into the structure-activity relationship of 1,7-naphthyridine inhibitors. This is relevant in that it provides information that can be invaluable as guidelines for the design of novel PIP4K2A inhibitors.
PIP4K2A是一种II型脂质激酶,催化磷脂酰肌醇-5-磷酸(PI5P)转化为磷脂酰肌醇4,5-二磷酸(PI4,5P2)的限速步骤。PIP4K2A与抑制各种类型肿瘤的活性氧介导的细胞凋亡密切相关,使其成为一个重要的治疗靶点。为了寻找具有高效PIP4K2A抑制活性的生物活性物质,使用机器学习算法研究了1,7-萘啶类似物的结构与抑制活性之间的定量关系。使用三种机器学习算法(MLR、ANN和SVM)开发了QSAR模型,该模型可以有效预测1,7-萘啶类似物文库的PIP4K2A抑制活性。通过依次应用GFA和MP5算法进行级联特征选择方法,以识别能够最好地描述1,7-萘啶类似物的PIP4K2A抑制活性的分子描述符子集。ML模型预测的PIP4K2A抑制活性与实验值高度相关。QSAR建模表明,性能最佳的ML模型是具有RBF核函数的SVM。SVM模型在预测1,7-萘啶类似物的PIP4K2A抑制活性方面表现非常出色,RTR和QEX值分别为0.9845和0.8793。为了进一步深入了解1,7-萘啶类似物的PIP4K2A抑制活性的结构起源,进行了分子对接研究。结果表明,发现五种化合物(15、25、13、09和28)与受体分子具有高结合亲和力。发现氢键、π-π相互作用和π-阳离子相互作用调节抑制剂的结合相互作用。尽管SVM本质上给出了一个难以解释的黑箱模型,但将SVM与MLR和ANN串联使用为构建强大的QSAR预测模型提供了独特的视角。ML模型的卓越预测性能和MLR模型的解释能力相结合,为1,7-萘啶抑制剂的构效关系提供了独特的见解。这很重要,因为它提供了作为新型PIP4K2A抑制剂设计指南可能非常宝贵的信息。