Penaud Benjamin, Laurent Benoit, Milhes Marine, Noüs Camille, Ehrenmann François, Dutech Cyril
BIOGECO, INRAE, Univ. Bordeaux, 33610 Cestas, France BIOGECO, INRAE, Univ. Bordeaux 33610 Cestas France.
INRAE, US 1426, GeT-PlaGe, Genotoul, Castanet-Tolosan, France INRAE, US 1426, GeT-PlaGe, Genotoul Castanet-Tolosan France.
Biodivers Data J. 2022 Aug 24;10:e85587. doi: 10.3897/BDJ.10.e85587. eCollection 2022.
For several decades, an increase in disease or pest emergences due to anthropogenic introduction or environmental changes has been recorded. This increase leads to serious threats to the genetic and species diversity of numerous ecosystems. Many of these events involve species with poor or no genomic resources (called here "orphan species"). This lack of resources is a serious limitation to our understanding of the origin of emergent populations, their ability to adapt to new environments and to predict future consequences to biodiversity. Analyses of genetic diversity are an efficient method to obtain this information rapidly, but require available polymorphic genetic markers.
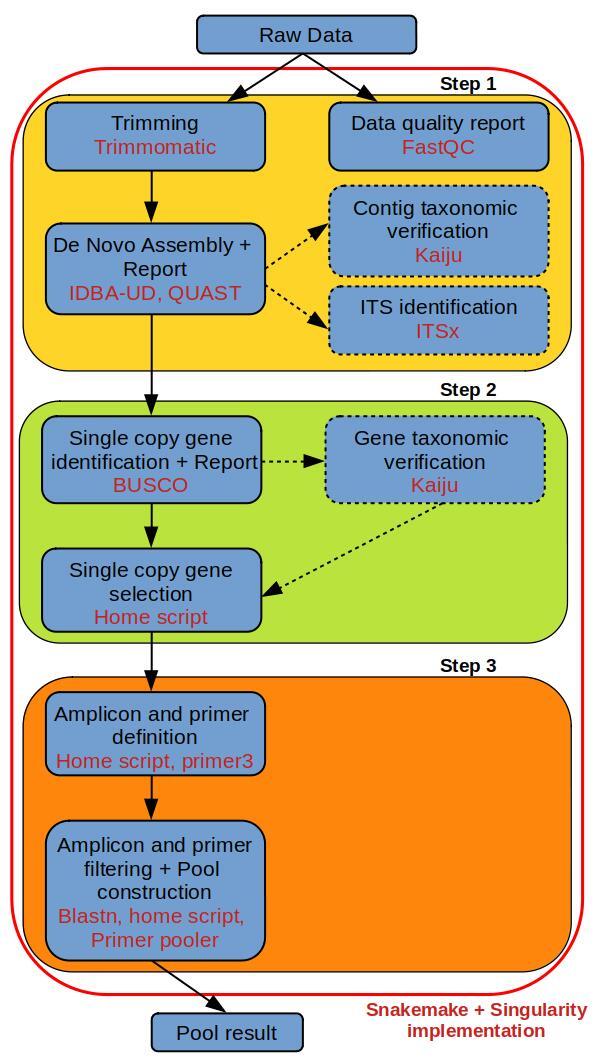
We developed a generic bioinformatics pipeline to rapidly isolate such markers with the goal for the pipeline to be applied in studies of invasive taxa from different taxonomic groups, with a special focus on forest fungal pathogens and insect pests. This pipeline is based on: 1) an automated de novo genome assembly obtained from shotgun whole genome sequencing using paired-end Illumina technology; 2) the isolation of single-copy genes conserved in species related to the studied emergent organisms; 3) primer development for multiplexed short sequences obtained from these conserved genes. Previous studies have shown that intronic regions of these conserved genes generally contain several single nucleotide polymorphisms within species. The pipeline's functionality was evaluated with sequenced genomes of five invasive or expanding pathogen and pest species in Europe ( (Romagn.) Herink 1973, Steiner & Buhrer 1934, (fr.) Dicko & B. Sutton 1980, (Griffon & Maubl.) U. Braun & S. Takam. 2000, Denis & Schiffermüller, 1775). We successfully isolated several pools of one hundred short gene regions for each assembled genome, which can be amplified in multiplex. The bioinformatics pipeline is user-friendly and requires little computational resources. This easy-to-set-up and run method for genetic marker identification will be useful for numerous laboratories studying biological invasions, but with limited resources and expertise in bioinformatics.
几十年来,因人为引入或环境变化导致的疾病或害虫出现事件不断增加。这种增加对众多生态系统的遗传和物种多样性构成了严重威胁。其中许多事件涉及基因组资源匮乏或没有基因组资源的物种(此处称为“孤儿物种”)。资源的匮乏严重限制了我们对新出现种群的起源、它们适应新环境的能力以及预测对生物多样性未来影响的理解。遗传多样性分析是快速获取此类信息的有效方法,但需要可用的多态性遗传标记。
我们开发了一种通用的生物信息学流程,以快速分离此类标记,目标是将该流程应用于不同分类群的入侵类群研究,特别关注森林真菌病原体和害虫。该流程基于:1)使用成对末端Illumina技术通过鸟枪法全基因组测序获得的自动从头基因组组装;2)在与所研究的新出现生物相关的物种中保守的单拷贝基因的分离;3)从这些保守基因获得的多重短序列的引物开发。先前的研究表明,这些保守基因的内含子区域通常在物种内包含多个单核苷酸多态性。该流程的功能通过对欧洲五种入侵或扩散的病原体和害虫物种((Romagn.) Herink 1973、Steiner & Buhrer 1934、(fr.) Dicko & B. Sutton 1980、(Griffon & Maubl.) U. Braun & S. Takam. 2000、Denis & Schiffermüller, 1775)的测序基因组进行了评估。我们成功地为每个组装基因组分离了几个包含一百个短基因区域的池,这些区域可以进行多重扩增。该生物信息学流程对用户友好,所需计算资源很少。这种易于设置和运行的遗传标记识别方法将对众多研究生物入侵但在生物信息学方面资源和专业知识有限的实验室有用。