Neuromuscular Unit, Department of Neurology, Hospital Sant Joan de Déu, Passeig Sant Joan de Déu 2, Esplugues de Llobregat, Barcelona, Spain.
Applied Research in Neuromuscular Diseases, Institut de Recerca Sant Joan de Déu, Barcelona, Spain.
Acta Neuropathol. 2023 Apr;145(4):479-496. doi: 10.1007/s00401-023-02551-7. Epub 2023 Feb 17.
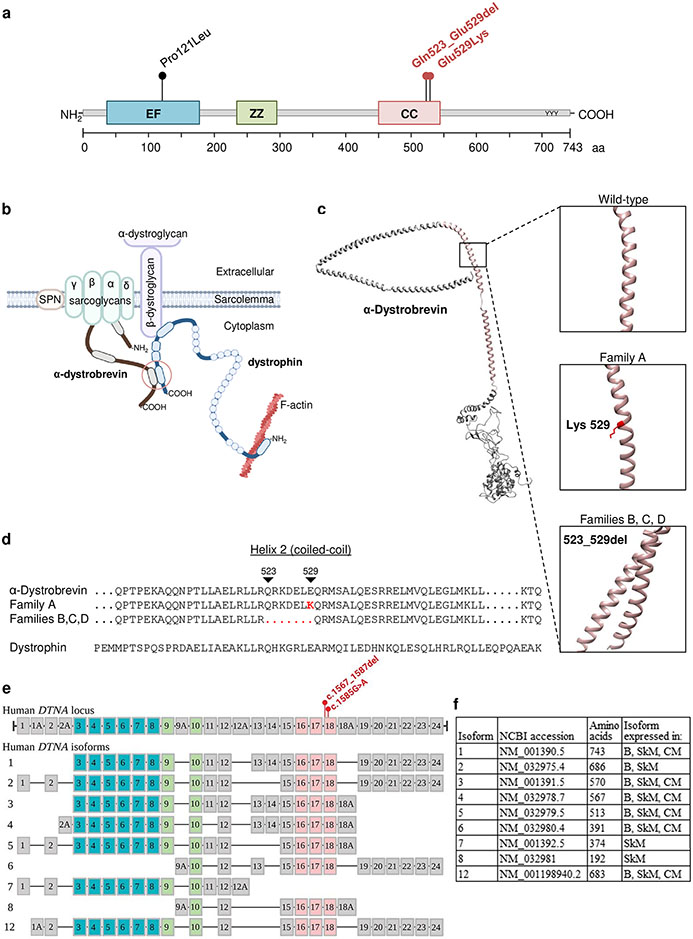
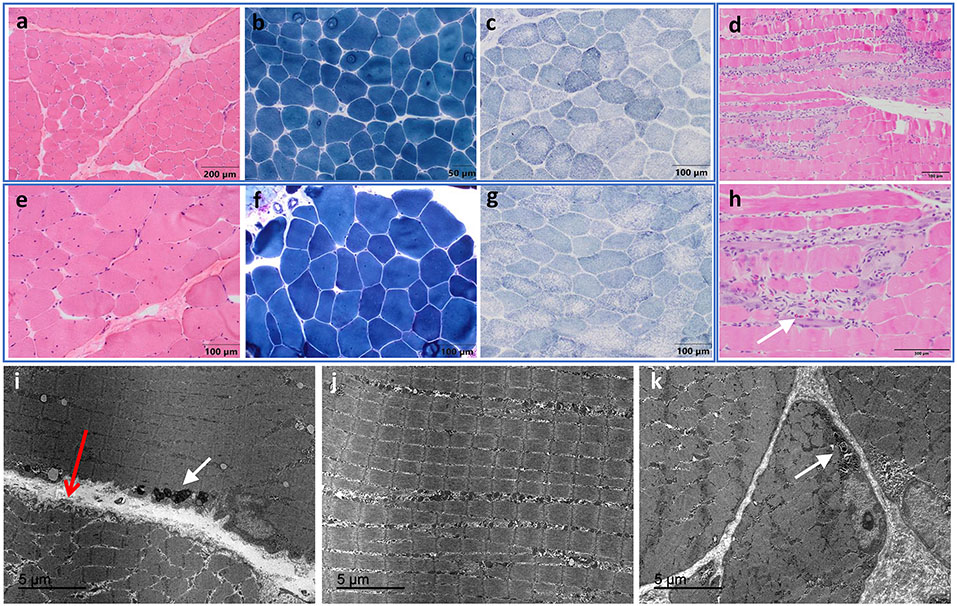
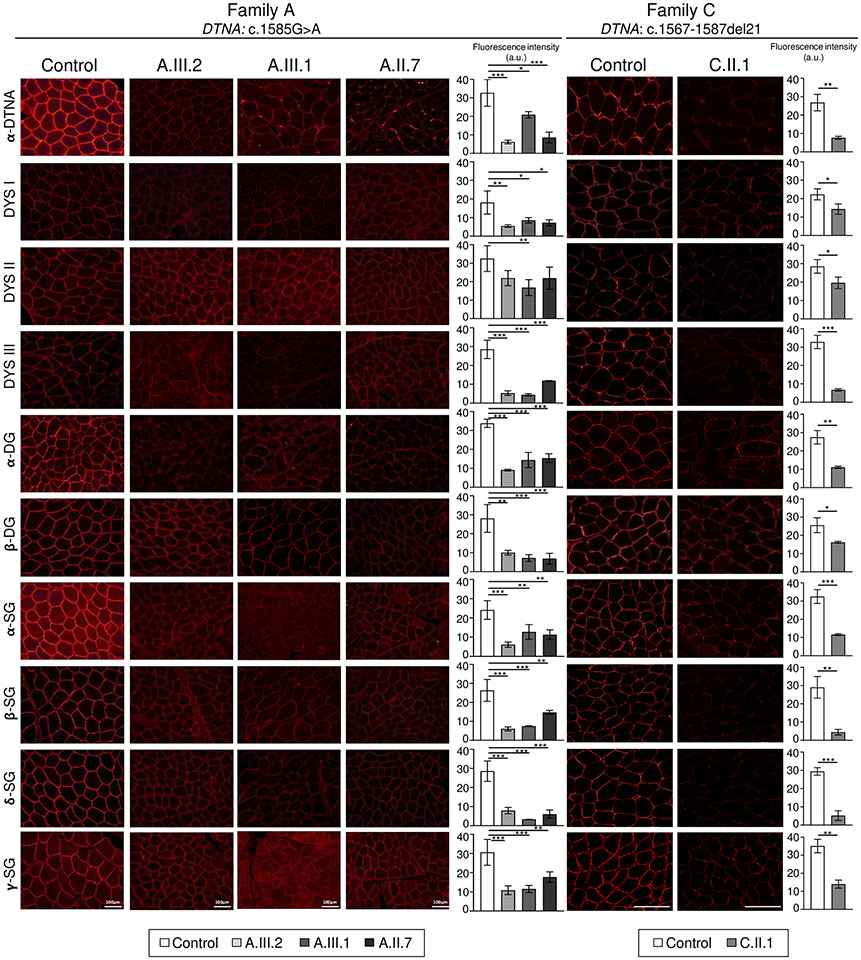
DTNA encodes α-dystrobrevin, a component of the macromolecular dystrophin-glycoprotein complex (DGC) that binds to dystrophin/utrophin and α-syntrophin. Mice lacking α-dystrobrevin have a muscular dystrophy phenotype, but variants in DTNA have not previously been associated with human skeletal muscle disease. We present 12 individuals from four unrelated families with two different monoallelic DTNA variants affecting the coiled-coil domain of α-dystrobrevin. The five affected individuals from family A harbor a c.1585G > A; p.Glu529Lys variant, while the recurrent c.1567_1587del; p.Gln523_Glu529del DTNA variant was identified in the other three families (family B: four affected individuals, family C: one affected individual, and family D: two affected individuals). Myalgia and exercise intolerance, with variable ages of onset, were reported in 10 of 12 affected individuals. Proximal lower limb weakness with onset in the first decade of life was noted in three individuals. Persistent elevations of serum creatine kinase (CK) levels were detected in 11 of 12 affected individuals, 1 of whom had an episode of rhabdomyolysis at 20 years of age. Autism spectrum disorder or learning disabilities were reported in four individuals with the c.1567_1587 deletion. Muscle biopsies in eight affected individuals showed mixed myopathic and dystrophic findings, characterized by fiber size variability, internalized nuclei, and slightly increased extracellular connective tissue and inflammation. Immunofluorescence analysis of biopsies from five affected individuals showed reduced α-dystrobrevin immunoreactivity and variably reduced immunoreactivity of other DGC proteins: dystrophin, α, β, δ and γ-sarcoglycans, and α and β-dystroglycans. The DTNA deletion disrupted an interaction between α-dystrobrevin and syntrophin. Specific variants in the coiled-coil domain of DTNA cause skeletal muscle disease with variable penetrance. Affected individuals show a spectrum of clinical manifestations, with severity ranging from hyperCKemia, myalgias, and exercise intolerance to childhood-onset proximal muscle weakness. Our findings expand the molecular etiologies of both muscular dystrophy and paucisymptomatic hyperCKemia, to now include monoallelic DTNA variants as a novel cause of skeletal muscle disease in humans.
DTNA 编码 α- dystrobrevin,这是一种大分子 dystrophin- glycoprotein 复合物(DGC)的组成部分,与 dystrophin/utrophin 和 α- syntrophin 结合。缺乏 α- dystrobrevin 的小鼠表现出肌肉营养不良表型,但 DTNA 的变异以前与人类骨骼肌疾病无关。我们介绍了来自四个无关家庭的 12 个人,他们都携带有两个不同的单等位基因 DTNA 变异,这些变异影响 α- dystrobrevin 的卷曲螺旋结构域。家族 A 的 5 名受影响个体携带有 c.1585G>A;p.Glu529Lys 变异,而在另外三个家族(家族 B:4 名受影响个体、家族 C:1 名受影响个体和家族 D:2 名受影响个体)中发现了重复出现的 c.1567_1587del;p.Gln523_Glu529del DTNA 变异。12 名受影响个体中有 10 名报告有肌痛和运动不耐受,发病年龄不同。3 名个体在生命的第一个十年出现了近端下肢无力。12 名受影响个体中有 11 名血清肌酸激酶(CK)水平持续升高,其中 1 名在 20 岁时发生过横纹肌溶解症。4 名携带有 c.1567_1587 缺失的个体报告有自闭症谱系障碍或学习障碍。8 名受影响个体的肌肉活检显示出混合肌病和营养不良的表现,特征为纤维大小变异、内化核、略微增加的细胞外结缔组织和炎症。对 5 名受影响个体的活检进行免疫荧光分析显示,α- dystrobrevin 免疫反应性降低,其他 DGC 蛋白的免疫反应性也不同程度降低:dystrophin、α、β、δ 和 γ- sarcoglycans,以及 α 和 β- dystroglycans。DTNA 缺失破坏了 α- dystrobrevin 与 syntrophin 之间的相互作用。DTNA 卷曲螺旋结构域的特定变异导致骨骼肌疾病,具有不同的外显率。受影响个体表现出一系列临床表现,严重程度从高 CK 血症、肌痛和运动不耐受到儿童期发病的近端肌肉无力不等。我们的发现扩展了肌肉营养不良症和无症状性高 CK 血症的分子病因学,现在将单等位基因 DTNA 变异作为人类骨骼肌疾病的新病因。