Jiang Dejun, Ye Zhaofeng, Hsieh Chang-Yu, Yang Ziyi, Zhang Xujun, Kang Yu, Du Hongyan, Wu Zhenxing, Wang Jike, Zeng Yundian, Zhang Haotian, Wang Xiaorui, Wang Mingyang, Yao Xiaojun, Zhang Shengyu, Wu Jian, Hou Tingjun
Innovation Institute for Artificial Intelligence in Medicine of Zhejiang University, College of Pharmaceutical Sciences, Zhejiang University Hangzhou 310058 Zhejiang China
Tencent Quantum Laboratory, Tencent Shenzhen 518057 Guangdong China
Chem Sci. 2023 Jan 19;14(8):2054-2069. doi: 10.1039/d2sc06576b. eCollection 2023 Feb 22.
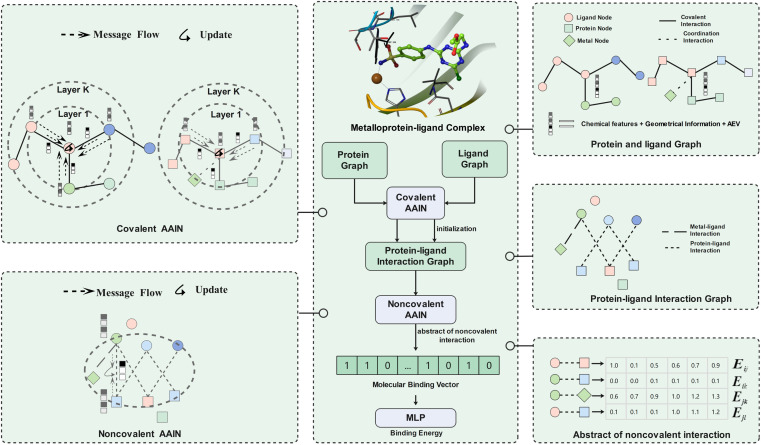
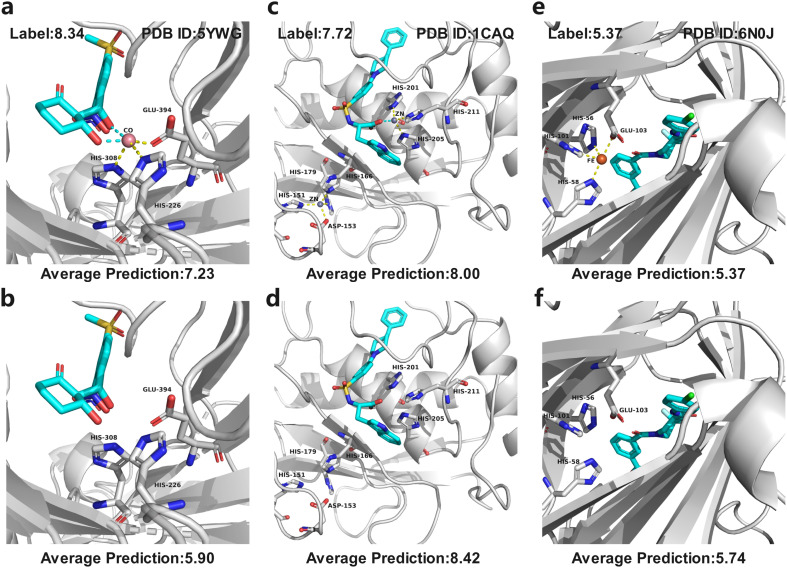
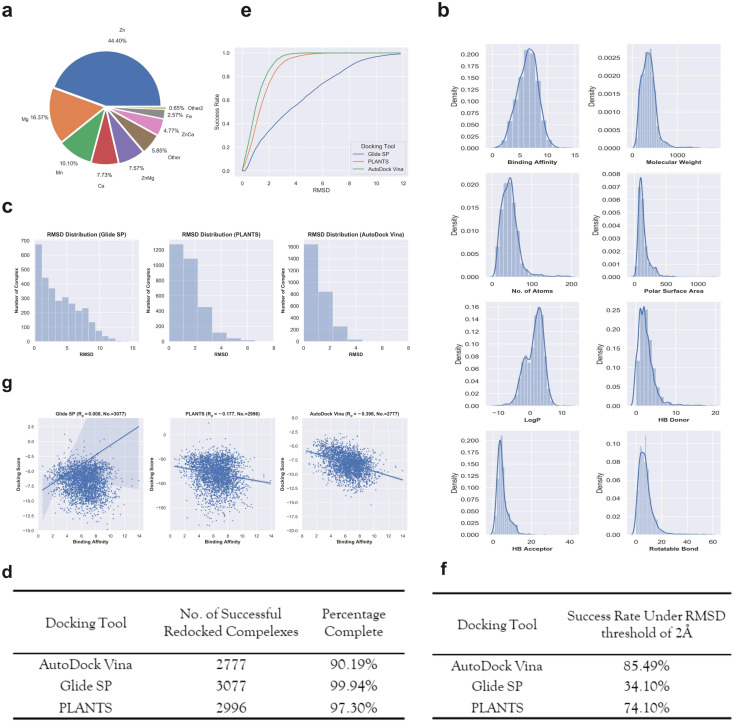
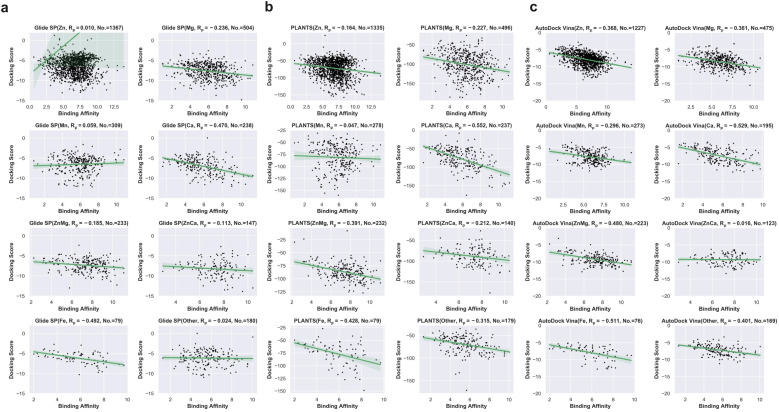
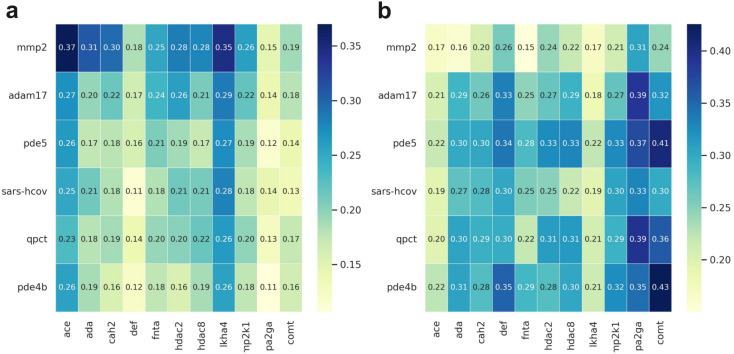
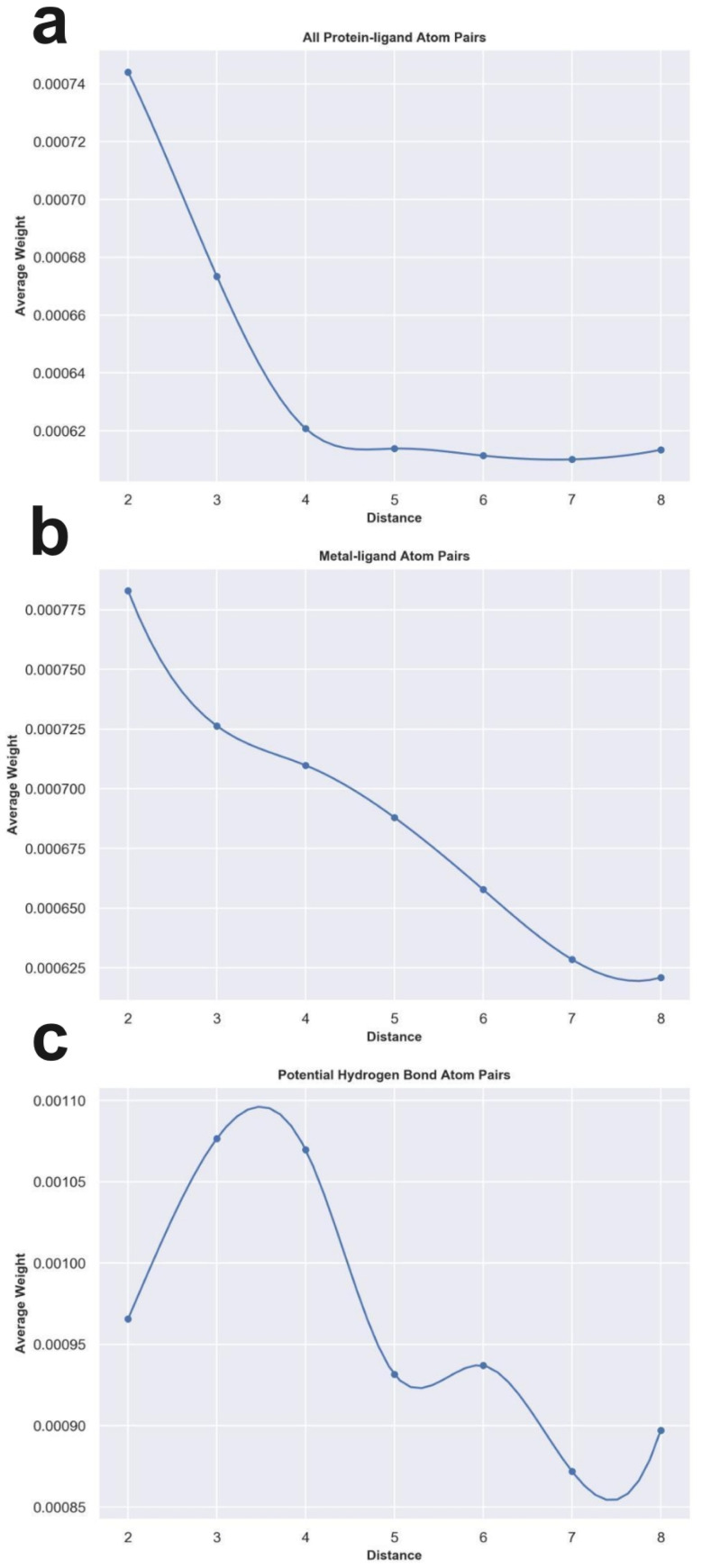
Metalloproteins play indispensable roles in various biological processes ranging from reaction catalysis to free radical scavenging, and they are also pertinent to numerous pathologies including cancer, HIV infection, neurodegeneration, and inflammation. Discovery of high-affinity ligands for metalloproteins powers the treatment of these pathologies. Extensive efforts have been made to develop approaches, such as molecular docking and machine learning (ML)-based models, for fast identification of ligands binding to heterogeneous proteins, but few of them have exclusively concentrated on metalloproteins. In this study, we first compiled the largest metalloprotein-ligand complex dataset containing 3079 high-quality structures, and systematically evaluated the scoring and docking powers of three competitive docking tools (, PLANTS, AutoDock Vina and Glide SP) for metalloproteins. Then, a structure-based deep graph model called MetalProGNet was developed to predict metalloprotein-ligand interactions. In the model, the coordination interactions between metal ions and protein atoms and the interactions between metal ions and ligand atoms were explicitly modelled through graph convolution. The binding features were then predicted by the informative molecular binding vector learned from a noncovalent atom-atom interaction network. The evaluation on the internal metalloprotein test set, the independent ChEMBL dataset towards 22 different metalloproteins and the virtual screening dataset indicated that MetalProGNet outperformed various baselines. Finally, a noncovalent atom-atom interaction masking technique was employed to interpret MetalProGNet, and the learned knowledge accords with our understanding of physics.
金属蛋白在从反应催化到自由基清除的各种生物过程中发挥着不可或缺的作用,并且它们还与包括癌症、艾滋病毒感染、神经退行性变和炎症在内的多种病理学相关。发现金属蛋白的高亲和力配体有助于治疗这些病理学疾病。人们已经做出了广泛的努力来开发各种方法,如分子对接和基于机器学习(ML)的模型,以快速识别与异质蛋白结合的配体,但其中很少有专门针对金属蛋白的。在本研究中,我们首先汇编了最大的金属蛋白 - 配体复合物数据集,其中包含3079个高质量结构,并系统地评估了三种竞争性对接工具(PLANTS、AutoDock Vina和Glide SP)对金属蛋白的评分和对接能力。然后,开发了一种基于结构的深度图模型MetalProGNet来预测金属蛋白 - 配体相互作用。在该模型中,通过图卷积明确地对金属离子与蛋白质原子之间的配位相互作用以及金属离子与配体原子之间的相互作用进行建模。然后通过从非共价原子 - 原子相互作用网络中学习到的信息性分子结合向量来预测结合特征。对内部金属蛋白测试集、针对22种不同金属蛋白的独立ChEMBL数据集以及虚拟筛选数据集的评估表明,MetalProGNet优于各种基线。最后,采用非共价原子 - 原子相互作用掩码技术来解释MetalProGNet,并且所学到的知识与我们对物理学的理解相符。