Skinner Kasey R, Koga Tomoyuki, Miki Shunichiro, Gruener Robert F, Grigore Florina-Nicoleta, Torii Emma H, Seelig Davis M, Suzuki Yuta, Kawauchi Daisuke, Lin Benjamin, Malicki Denise M, Chen Clark C, Benveniste Etty N, Patel Rakesh P, McFarland Braden C, Huang R Stephanie, Jones Chris, Mackay Alan, Miller C Ryan, Furnari Frank B
Division of Neuropathology, Department of Pathology, O'Neal Comprehensive Cancer Center and Comprehensive Neuroscience Center, Heersink School of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Neuroscience Curriculum, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
bioRxiv. 2023 Feb 24:2023.02.24.528982. doi: 10.1101/2023.02.24.528982.
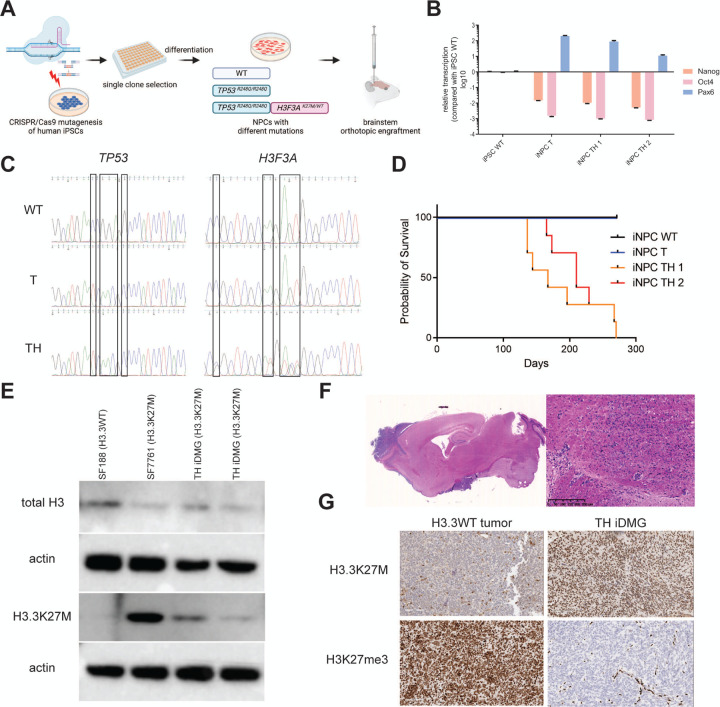
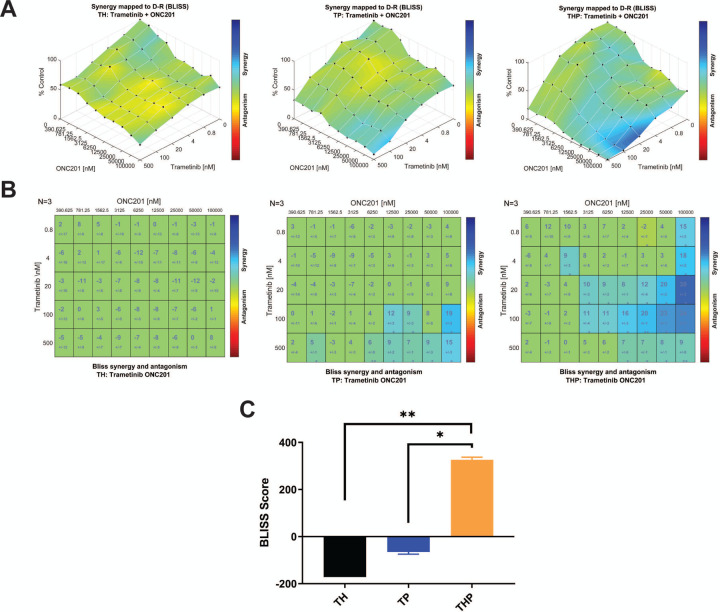
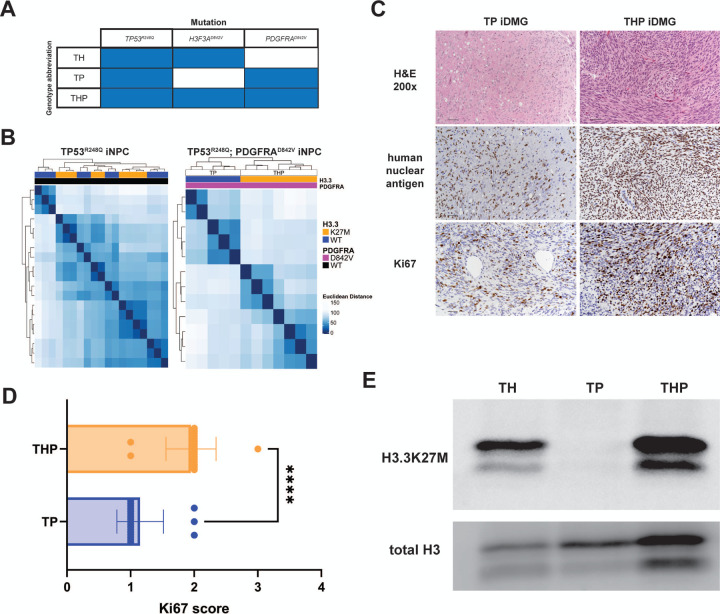
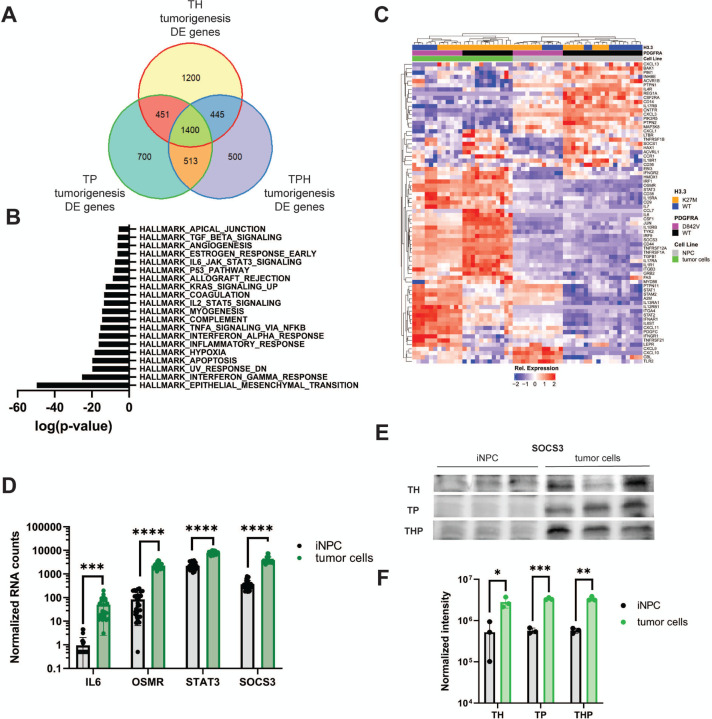
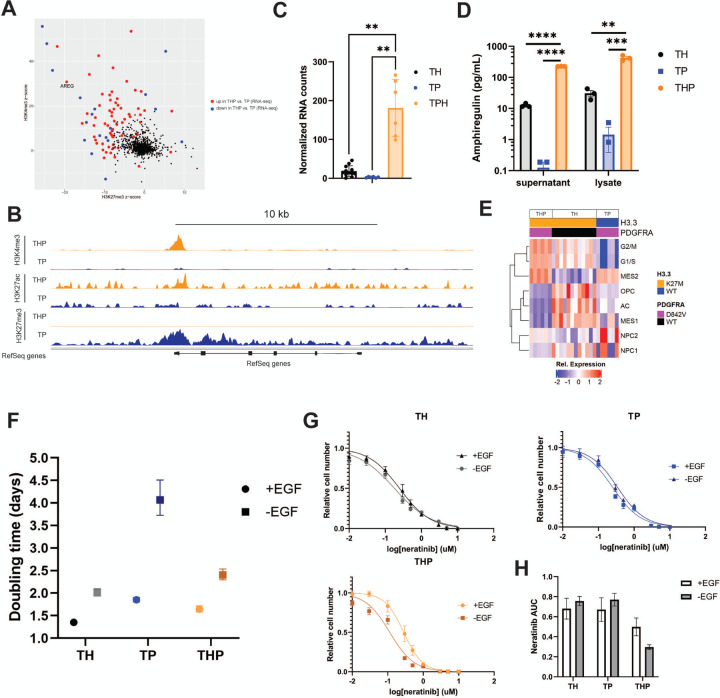
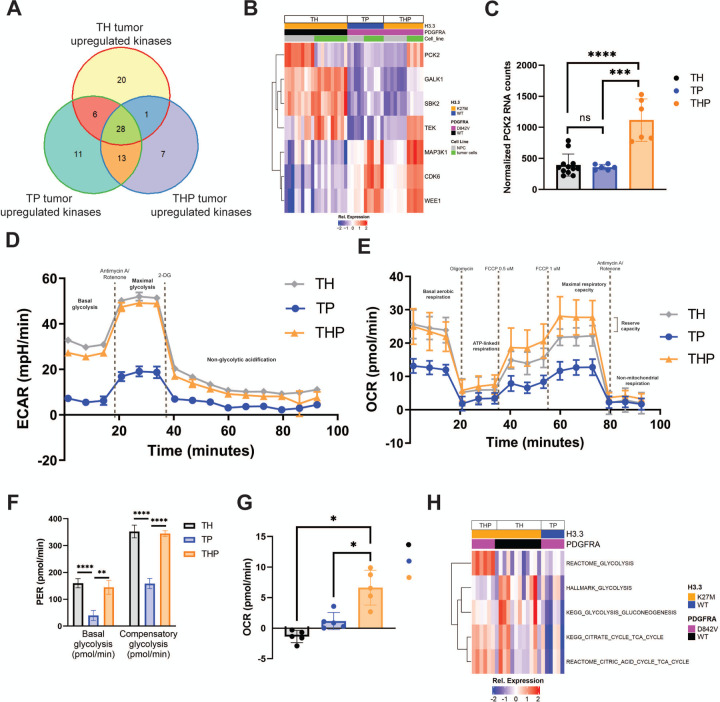
Diffuse midline glioma (DMG) is a leading cause of brain tumor death in children. In addition to hallmark H3.3K27M mutations, significant subsets also harbor alterations of other genes, such as and . Despite the prevalence of H3.3K27M, the results of clinical trials in DMG have been mixed, possibly due to the lack of models recapitulating its genetic heterogeneity. To address this gap, we developed human iPSC-derived tumor models harboring TP53 with or without heterozygous H3.3K27M and/or PDGFRA overexpression. The combination of H3.3K27M and PDGFRA resulted in more proliferative tumors when gene-edited neural progenitor (NP) cells were implanted into mouse brains compared to NP with either mutation alone. Transcriptomic comparison of tumors and their NP cells of origin identified conserved JAK/STAT pathway activation across genotypes as characteristic of malignant transformation. Conversely, integrated genome-wide epigenomic and transcriptomic analyses, as well as rational pharmacologic inhibition, revealed targetable vulnerabilities unique to the TP53; H3.3K27M; PDGFRA tumors and related to their aggressive growth phenotype. These include -mediated cell cycle control, altered metabolism, and vulnerability to combination ONC201/trametinib treatment. Taken together, these data suggest that cooperation between H3.3K27M and PDGFRA influences tumor biology, underscoring the need for better molecular stratification in DMG clinical trials.
弥漫性中线胶质瘤(DMG)是儿童脑肿瘤死亡的主要原因。除了标志性的H3.3K27M突变外,相当一部分肿瘤还存在其他基因的改变,如 和 。尽管H3.3K27M普遍存在,但DMG临床试验的结果却喜忧参半,这可能是由于缺乏能够概括其基因异质性的模型。为了填补这一空白,我们构建了携带TP53且有或没有杂合H3.3K27M和/或PDGFRA过表达的人诱导多能干细胞衍生肿瘤模型。当将基因编辑的神经祖细胞(NP)植入小鼠脑内时,与单独携带任一突变的NP相比,H3.3K27M和PDGFRA共同作用导致肿瘤增殖性更强。对肿瘤及其来源的NP细胞进行转录组比较,发现不同基因型中保守的JAK/STAT通路激活是恶性转化的特征。相反,全基因组表观基因组和转录组的综合分析以及合理的药物抑制,揭示了TP53、H3.3K27M、PDGFRA肿瘤特有的、与其侵袭性生长表型相关的可靶向弱点。这些弱点包括 介导的细胞周期控制、代谢改变以及对ONC201/曲美替尼联合治疗的敏感性。综上所述,这些数据表明H3.3K27M和PDGFRA之间的协同作用影响肿瘤生物学特性,强调了在DMG临床试验中进行更好的分子分层的必要性。