Ringman John M, Dorrani Naghmeh, Fernández Sara Gutiérrez, Signer Rebecca, Martinez-Agosto Julian, Lee Hane, Douine Emilie D, Qiao Yuchuan, Shi Yonggang, D'Orazio Lina, Pawar Sanjay, Robbie Leah, Kashani Amir H, Singer Maxwell, Byers Joshua T, Magaki Shino, Guzman Sam, Sagare Abhay, Zlokovic Berislav, Cederbaum Stephen, Nelson Stanley, Sheikh-Bahaei Nasim, Chui Helena C, Chávez-Gutiérrez Lucía, Vinters Harry V
Department of Neurology, Keck School of Medicine at University of Southern California, Los Angeles, CA 90033, USA.
Department of Pediatrics, UCLA, Los Angeles, CA 90095, USA.
Brain Commun. 2023 Feb 15;5(2):fcad030. doi: 10.1093/braincomms/fcad030. eCollection 2023.
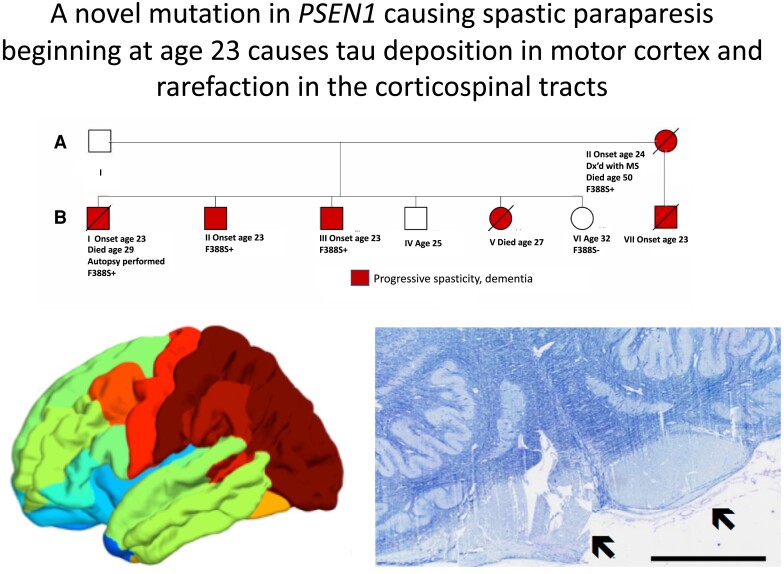
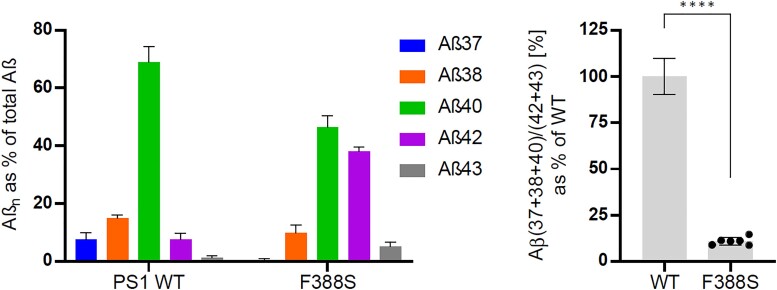
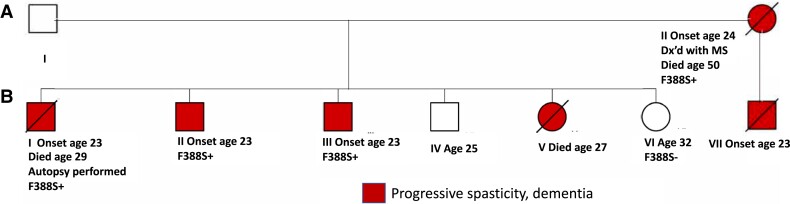
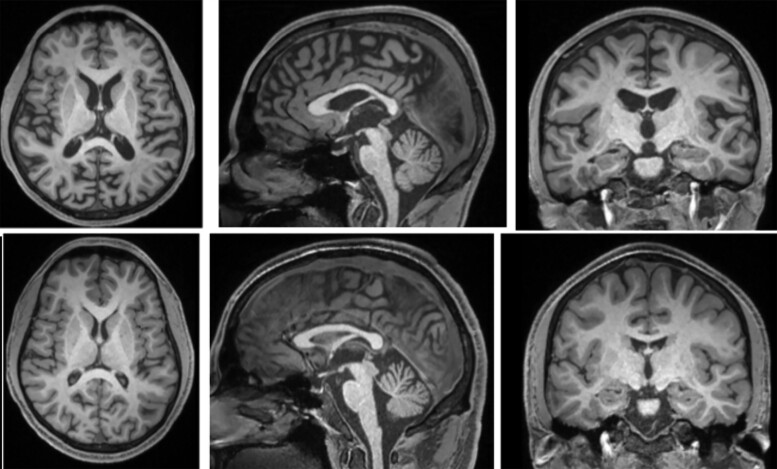
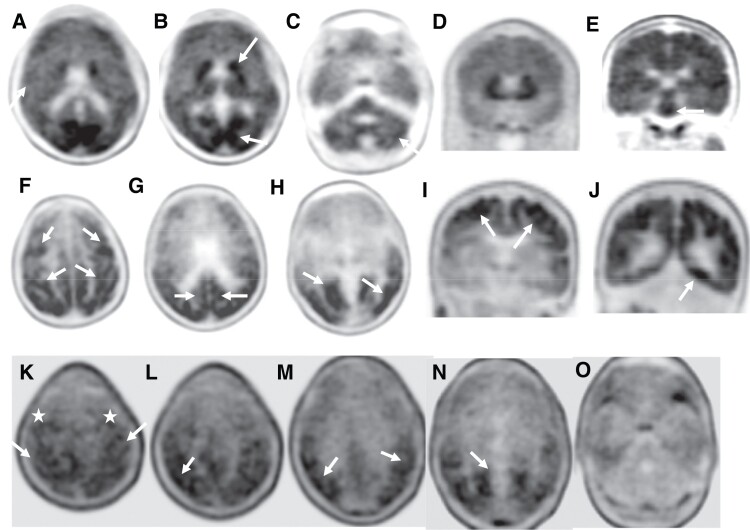
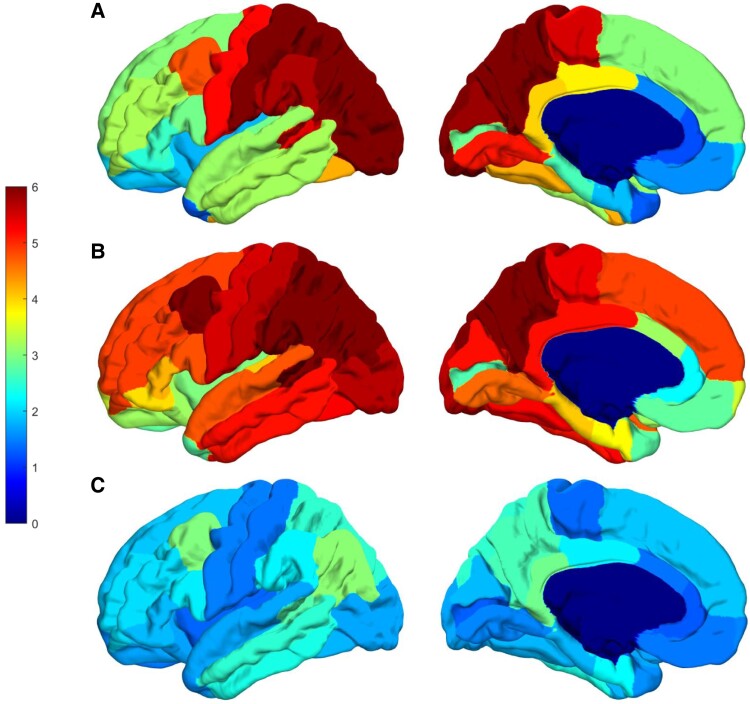
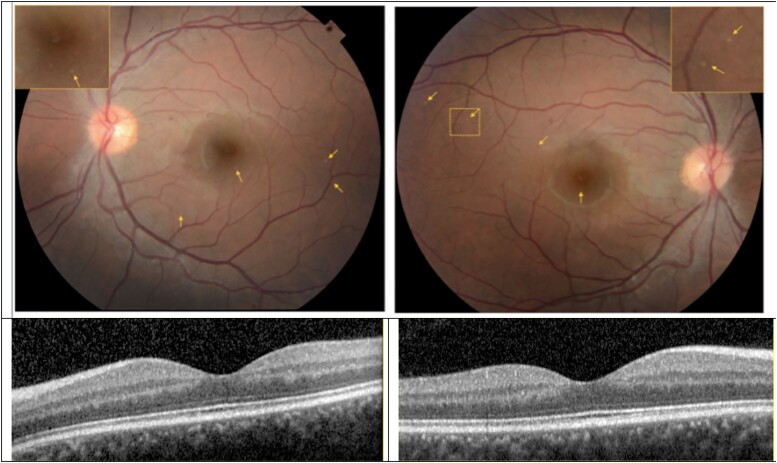
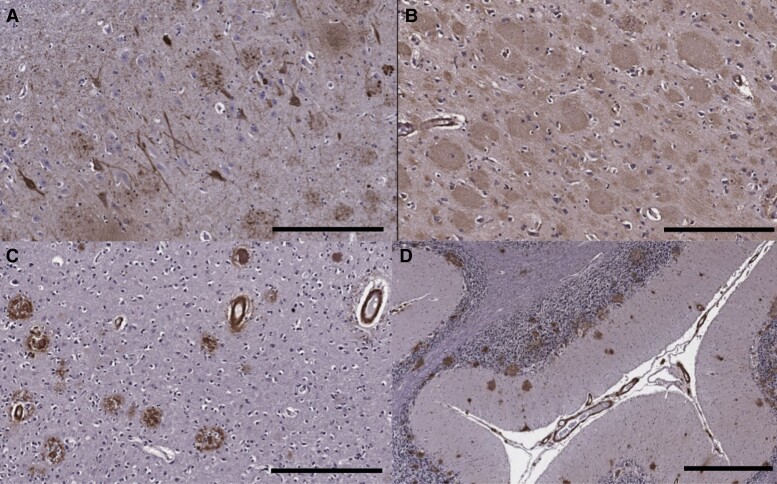
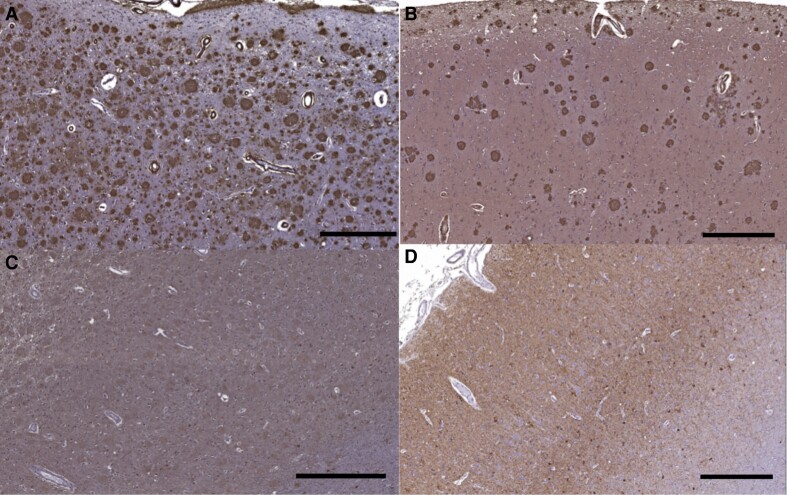
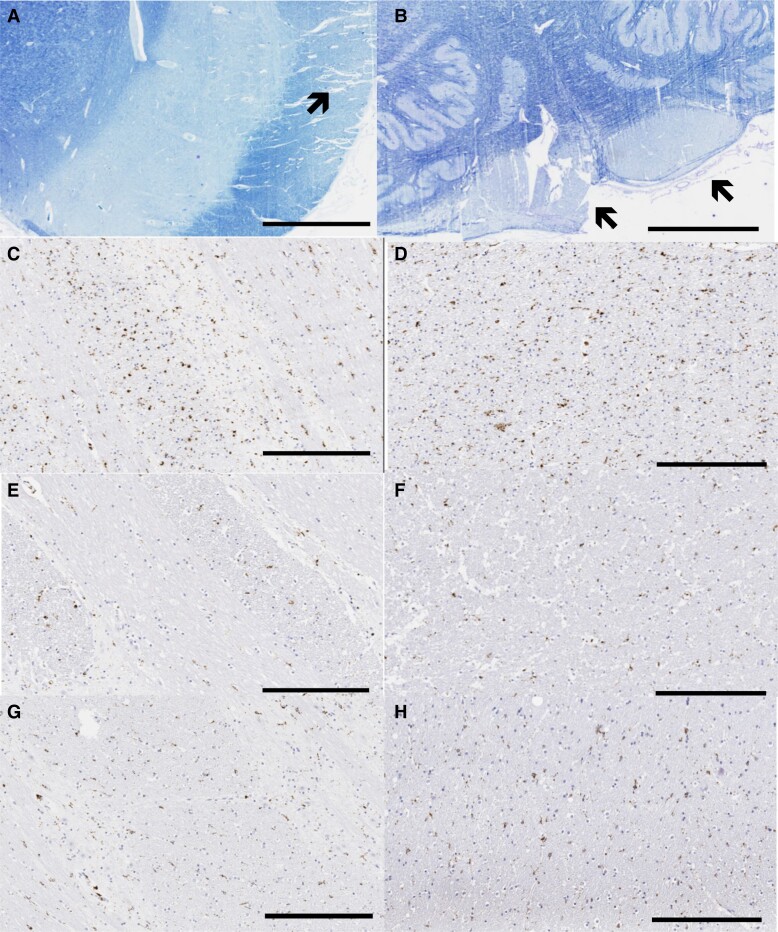
Spastic paraparesis has been described to occur in 13.7% of mutations and can be the presenting feature in 7.5%. In this paper, we describe a family with a particularly young onset of spastic paraparesis due to a novel mutation in (F388S). Three affected brothers underwent comprehensive imaging protocols, two underwent ophthalmological evaluations and one underwent neuropathological examination after his death at age 29. Age of onset was consistently at age 23 with spastic paraparesis, dysarthria and bradyphrenia. Pseudobulbar affect followed with progressive gait problems leading to loss of ambulation in the late 20s. Cerebrospinal fluid levels of amyloid-β, tau and phosphorylated tau and florbetaben PET were consistent with Alzheimer's disease. Flortaucipir PET showed an uptake pattern atypical for Alzheimer's disease, with disproportionate signal in posterior brain areas. Diffusion tensor imaging showed decreased mean diffusivity in widespread areas of white matter but particularly in areas underlying the peri-Rolandic cortex and in the corticospinal tracts. These changes were more severe than those found in carriers of another mutation, which can cause spastic paraparesis at a later age (A431E), which were in turn more severe than among persons carrying autosomal dominant Alzheimer's disease mutations not causing spastic paraparesis. Neuropathological examination confirmed the presence of cotton wool plaques previously described in association with spastic parapresis and pallor and microgliosis in the corticospinal tract with severe amyloid-β pathology in motor cortex but without unequivocal disproportionate neuronal loss or tau pathology. modelling of the effects of the mutation demonstrated increased production of longer length amyloid-β peptides relative to shorter that predicted the young age of onset. In this paper, we provide imaging and neuropathological characterization of an extreme form of spastic paraparesis occurring in association with autosomal dominant Alzheimer's disease, demonstrating robust diffusion and pathological abnormalities in white matter. That the amyloid-β profiles produced predicted the young age of onset suggests an amyloid-driven aetiology though the link between this and the white matter pathology remains undefined.
痉挛性截瘫在13.7%的突变中被描述为会发生,且在7.5%的病例中可能是首发特征。在本文中,我们描述了一个因(F388S)基因的新突变而导致痉挛性截瘫发病特别早的家族。三名患病兄弟接受了全面的影像学检查,两名接受了眼科评估,一名在29岁去世后接受了神经病理学检查。发病年龄始终为23岁,伴有痉挛性截瘫、构音障碍和思维迟缓。随后出现假性延髓麻痹,并伴有进行性步态问题,导致在20多岁后期无法行走。脑脊液中β淀粉样蛋白、tau蛋白和磷酸化tau蛋白水平以及氟比他班PET检查结果与阿尔茨海默病一致。氟代脱氧葡萄糖PET显示出一种与阿尔茨海默病不同的摄取模式,在后脑区域信号不成比例。扩散张量成像显示,广泛的白质区域平均扩散率降低,尤其是在罗兰多周围皮质下方区域和皮质脊髓束中。这些变化比另一种可在较晚年龄导致痉挛性截瘫的突变(A431E)携带者中发现的变化更严重,而后者又比携带不导致痉挛性截瘫的常染色体显性阿尔茨海默病突变的人更严重。神经病理学检查证实存在先前描述的与痉挛性截瘫相关的棉絮状斑块,以及皮质脊髓束中的苍白和小胶质细胞增生,运动皮质存在严重的β淀粉样蛋白病理改变,但没有明确的不成比例的神经元丢失或tau蛋白病理改变。对该突变影响的建模显示,相对于较短的β淀粉样蛋白肽,较长长度的β淀粉样蛋白肽产生增加,这预测了发病年龄较早。在本文中,我们提供了与常染色体显性阿尔茨海默病相关的一种极端形式的痉挛性截瘫的影像学和神经病理学特征,证明了白质中存在明显的扩散和病理异常。所产生的β淀粉样蛋白谱预测了发病年龄较早,这表明存在由β淀粉样蛋白驱动的病因,尽管这与白质病理之间的联系尚不清楚。