Chelban Viorica, Breza Marianthi, Szaruga Maria, Vandrovcova Jana, Murphy David, Lee Chia-Ju, Alikhwan Sondos, Bourinaris Thomas, Vavougios George, Ilyas Muhammad, Halim Sobia Ahsan, Al-Harrasi Ahmed, Kartanou Chrisoula, Ronald Coras, Blumcke Ingmar, Alexoudi Athanasia, Gatzonis Stylianos, Stefanis Leonidas, Karadima Georgia, Wood Nicholas W, Chávez-Gutiérrez Lucía, Hardy John, Houlden Henry, Koutsis Georgios
Department of Neuromuscular Disease, Queen Square Institute of Neurology University College London London UK.
Department of Neurology and Neurosurgery Institute of Emergency Medicine Toma Ciorbă 1 Chisinau Republic of Moldova.
Alzheimers Dement (Amst). 2021 May 2;13(1):e12186. doi: 10.1002/dad2.12186. eCollection 2021.
We investigated the frequency, neuropathology, and phenotypic characteristics of spastic paraplegia (SP) that precedes dementia in presenilin 1 () related familial Alzheimer's disease (AD).
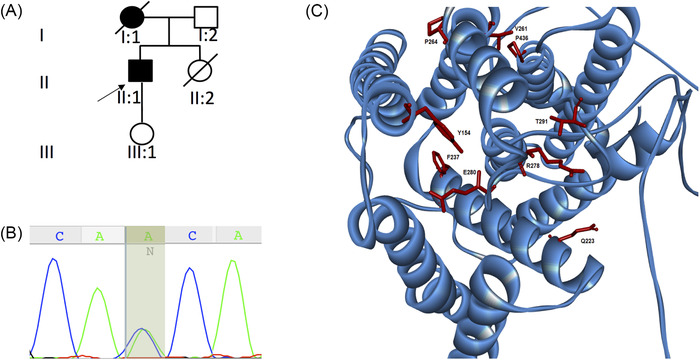
We performed whole exome sequencing (WES) in 60 probands with hereditary spastic paraplegia (HSP) phenotype that was negative for variants in known HSP-related genes. Where mutation was identified, brain biopsy was performed. We investigated the link between HSP and AD with PSEN1 in silico pathway analysis and measured the stability of PSEN1 mutant γ-secretase.
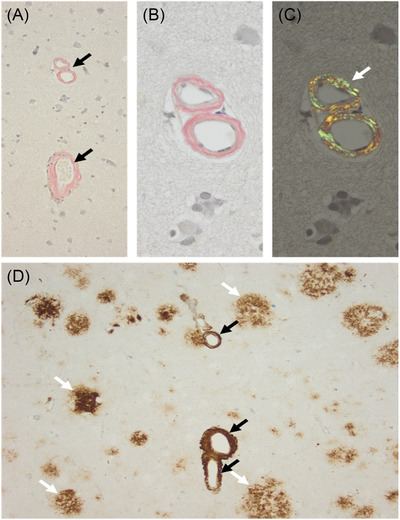
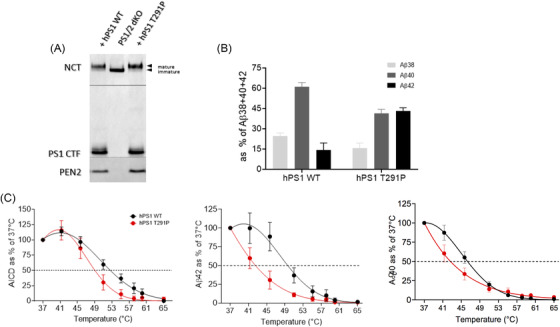
We identified a variant (p.Thr291Pro) in an individual presenting with pure SP at 30 years of age. Three years later, SP was associated with severe, fast cognitive decline and amyloid deposition with diffuse cortical plaques on brain biopsy. Biochemical analysis of p.Thr291Pro revealed that although the mutation does not alter active γ-secretase reconstitution, it destabilizes γ-secretase-amyloid precursor protein (APP)/amyloid beta (Aβn) interactions during proteolysis, enhancing the production of longer Aβ peptides. We then extended our analysis to all 226 pathogenic variants reported and show that 7.5% were associated with pure SP onset followed by cognitive decline later in the disease. We found that cases manifesting initially as SP have a later age of onset, are associated with mutations located beyond codon 200, and showed larger diffuse, cored plaques, amyloid-ring arteries, and severe CAA.
We show that pure SP can precede dementia onset in related familial AD. We recommend genetic testing in patients presenting with SP with no variants in known HSP-related genes, particularly when associated with a family history of cognitive decline.
我们研究了早老素1(PSEN1)相关家族性阿尔茨海默病(AD)中痴呆前痉挛性截瘫(SP)的频率、神经病理学和表型特征。
我们对60例具有遗传性痉挛性截瘫(HSP)表型且已知HSP相关基因变异呈阴性的先证者进行了全外显子组测序(WES)。在鉴定出PSEN1突变的情况下,进行了脑活检。我们通过计算机通路分析研究了HSP与PSEN1相关AD之间的联系,并测量了PSEN1突变型γ-分泌酶的稳定性。
我们在一名30岁时出现单纯性SP的个体中鉴定出一个PSEN1变异(p.Thr291Pro)。三年后,SP与严重、快速的认知衰退以及脑活检时弥漫性皮质斑块的淀粉样蛋白沉积相关。对p.Thr291Pro PSEN1的生化分析表明,尽管该突变不会改变活性γ-分泌酶的重组,但它会破坏蛋白水解过程中γ-分泌酶-淀粉样前体蛋白(APP)/淀粉样β(Aβn)的相互作用,从而增加更长Aβ肽的产生。然后,我们将分析扩展到所有报道的226个PSEN1致病变异,发现7.5%与单纯性SP起病相关,随后在疾病后期出现认知衰退。我们发现,最初表现为SP的病例发病年龄较晚,与密码子200以外的突变相关,并且显示出更大的弥漫性、有核心的斑块、淀粉样环动脉和严重的脑淀粉样血管病(CAA)。
我们表明,在PSEN1相关家族性AD中,单纯性SP可先于痴呆发病。我们建议对已知HSP相关基因无变异的SP患者进行基因检测,特别是当伴有认知衰退家族史时。