Kumar Lavanya, Leko Katarina, Nemec Vinko, Trzybiński Damian, Bregović Nikola, Cinčić Dominik, Arhangelskis Mihails
Faculty of Chemistry, University of Warsaw 1 Pasteura St. 02-093 Warsaw Poland
Faculty of Science, Department of Chemistry, University of Zagreb Horvatovac 102a HR-10000 Zagreb Croatia.
Chem Sci. 2023 Feb 8;14(12):3140-3146. doi: 10.1039/d2sc06770f. eCollection 2023 Mar 22.
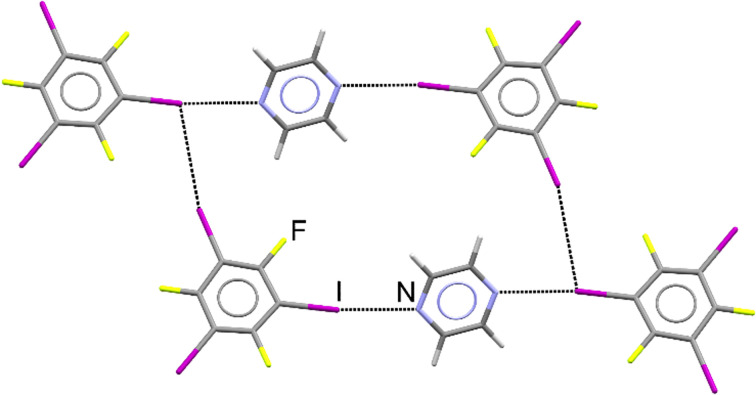
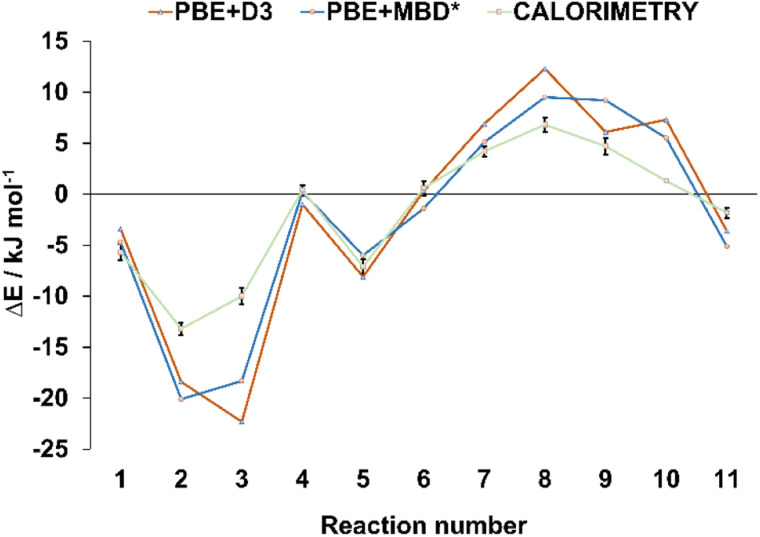
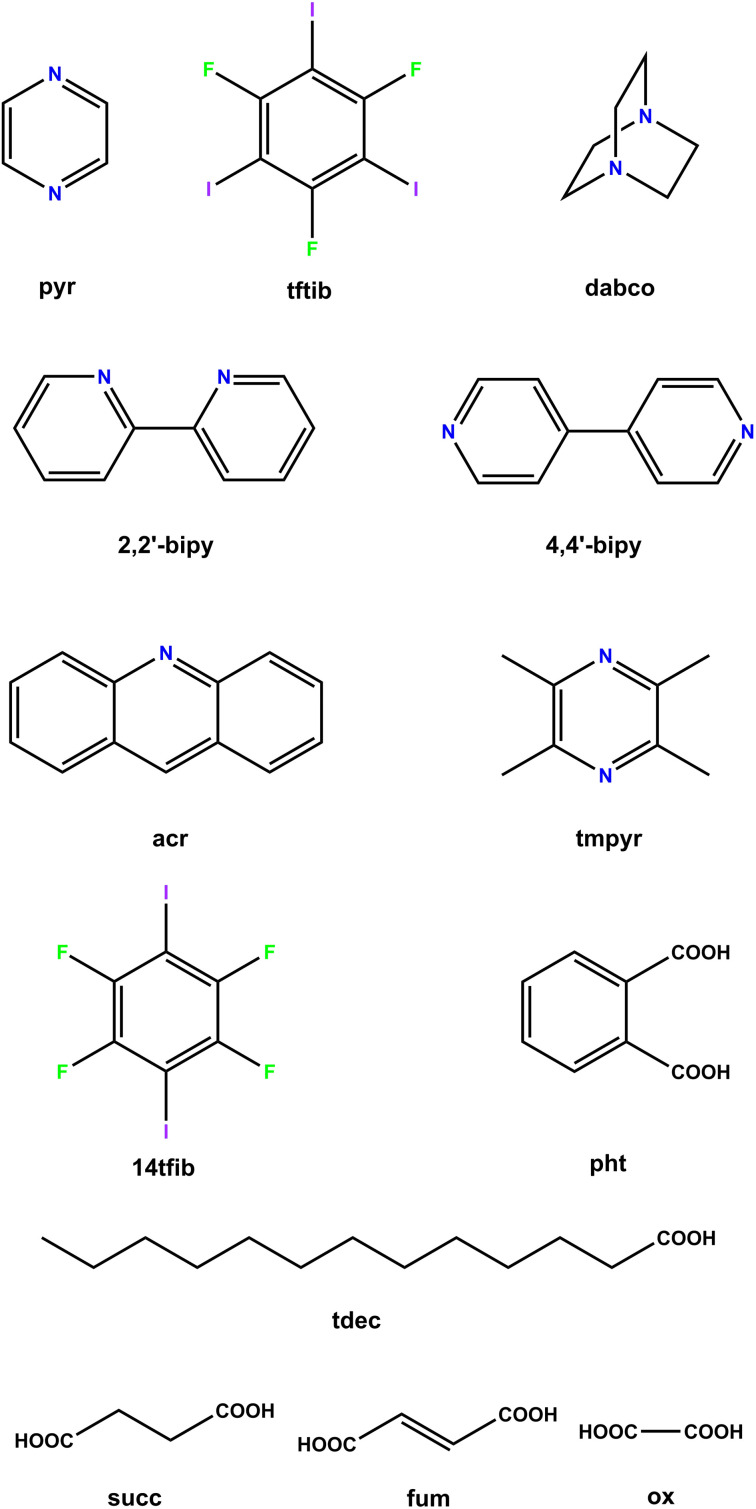
Periodic density-functional theory (DFT) calculations were used to predict the thermodynamic stability and the likelihood of interconversion between a series of halogen-bonded cocrystals. The outcomes of mechanochemical transformations were in excellent agreement with the theoretical predictions, demonstrating the power of periodic DFT as a method for designing solid-state mechanochemical reactions prior to experimental work. Furthermore, the calculated DFT energies were compared with experimental dissolution calorimetry measurements, marking the first such benchmark for the accuracy of periodic DFT calculations in modelling transformations of halogen-bonded molecular crystals.
采用周期性密度泛函理论(DFT)计算来预测一系列卤素键合共晶体之间的热力学稳定性和相互转化的可能性。机械化学转化的结果与理论预测高度吻合,证明了周期性DFT作为一种在实验工作之前设计固态机械化学反应的方法的强大功能。此外,将计算得到的DFT能量与实验溶解量热法测量结果进行了比较,这是首次对周期性DFT计算在模拟卤素键合分子晶体转化方面的准确性进行此类基准测试。