Department of Psychological & Brain Sciences, Washington University in St. Louis Medical School, Saint Louis, Missouri, USA.
Department of Psychiatry, Washington University School of Medicine, Saint Louis, Missouri, USA.
Genes Brain Behav. 2023 Dec;22(6):e12846. doi: 10.1111/gbb.12846. Epub 2023 Mar 28.
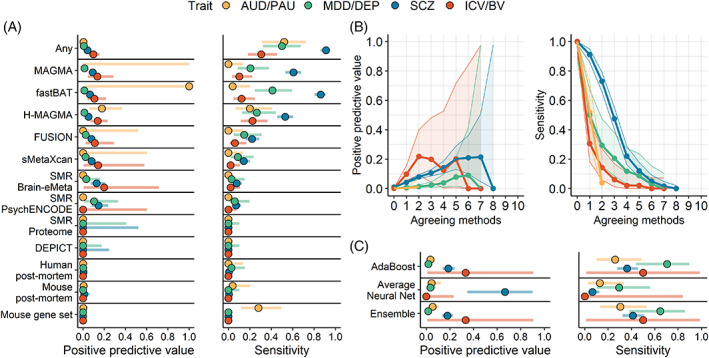
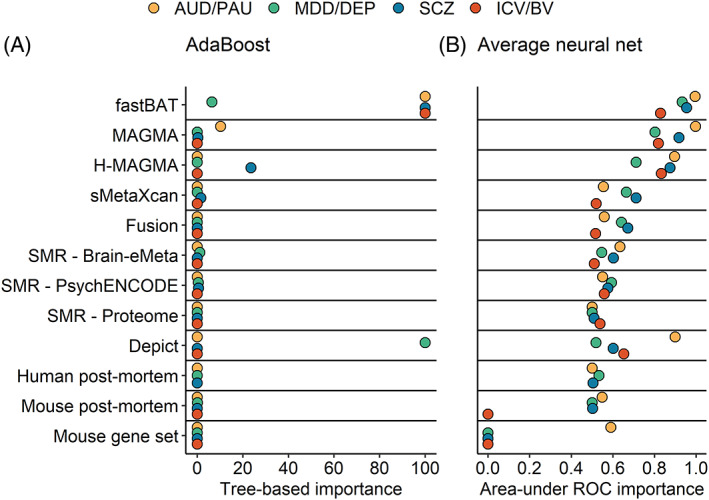
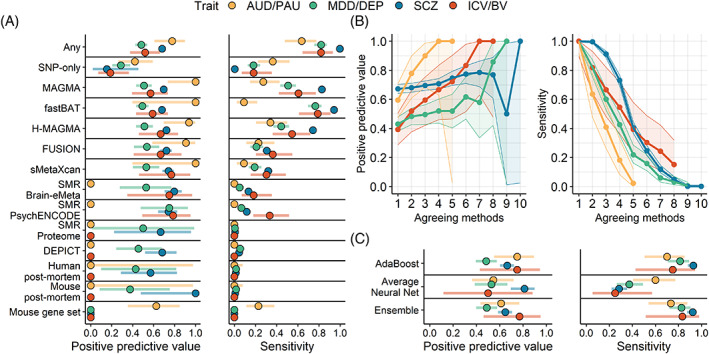
The integration of multi-omics information (e.g., epigenetics and transcriptomics) can be useful for interpreting findings from genome-wide association studies (GWAS). It has been suggested that multi-omics could circumvent or greatly reduce the need to increase GWAS sample sizes for novel variant discovery. We tested whether incorporating multi-omics information in earlier and smaller-sized GWAS boosts true-positive discovery of genes that were later revealed by larger GWAS of the same/similar traits. We applied 10 different analytic approaches to integrating multi-omics data from 12 sources (e.g., Genotype-Tissue Expression project) to test whether earlier and smaller GWAS of 4 brain-related traits (alcohol use disorder/problematic alcohol use, major depression/depression, schizophrenia, and intracranial volume/brain volume) could detect genes that were revealed by a later and larger GWAS. Multi-omics data did not reliably identify novel genes in earlier less-powered GWAS (PPV <0.2; 80% false-positive associations). Machine learning predictions marginally increased the number of identified novel genes, correctly identifying 1-8 additional genes, but only for well-powered early GWAS of highly heritable traits (i.e., intracranial volume and schizophrenia). Although multi-omics, particularly positional mapping (i.e., fastBAT, MAGMA, and H-MAGMA), can help to prioritize genes within genome-wide significant loci (PPVs = 0.5-1.0) and translate them into information about disease biology, it does not reliably increase novel gene discovery in brain-related GWAS. To increase power for discovery of novel genes and loci, increasing sample size is required.
多组学信息(例如,表观遗传学和转录组学)的整合对于解释全基因组关联研究(GWAS)的结果可能是有用的。有人认为,多组学可以避免或大大减少为发现新变体而增加 GWAS 样本量的必要性。我们测试了在早期和较小规模的 GWAS 中纳入多组学信息是否可以提高后来通过相同/相似特征的更大 GWAS 揭示的基因的真正阳性发现。我们应用了 10 种不同的分析方法,整合了来自 12 个来源的多组学数据(例如,基因型-组织表达项目),以测试 4 个与大脑相关的特征(酒精使用障碍/问题性饮酒、重度抑郁症/抑郁症、精神分裂症和颅内体积/脑容量)的早期和较小规模的 GWAS 是否可以检测到后来更大规模的 GWAS 揭示的基因。多组学数据无法可靠地识别早期、功率较低的 GWAS 中的新基因(PPV<0.2;80%的假阳性关联)。机器学习预测略微增加了识别的新基因数量,正确识别了 1-8 个额外的基因,但仅适用于具有高度遗传性特征的早期、功率较高的 GWAS(即颅内体积和精神分裂症)。尽管多组学,特别是定位映射(即 fastBAT、MAGMA 和 H-MAGMA),可以帮助在全基因组显著位点(PPV=0.5-1.0)中优先考虑基因,并将其转化为有关疾病生物学的信息,但它并不能可靠地增加与大脑相关的 GWAS 中新基因的发现。为了增加发现新基因和位点的能力,需要增加样本量。