Sasse Alexander, Ng Bernard, Spiro Anna E, Tasaki Shinya, Bennett David A, Gaiteri Christopher, De Jager Philip L, Chikina Maria, Mostafavi Sara
Paul G. Allen School of Computer Science and Engineering, University of Washington, WA, USA, 98195.
Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago, Illinois, USA, 60612.
bioRxiv. 2023 Sep 28:2023.03.16.532969. doi: 10.1101/2023.03.16.532969.
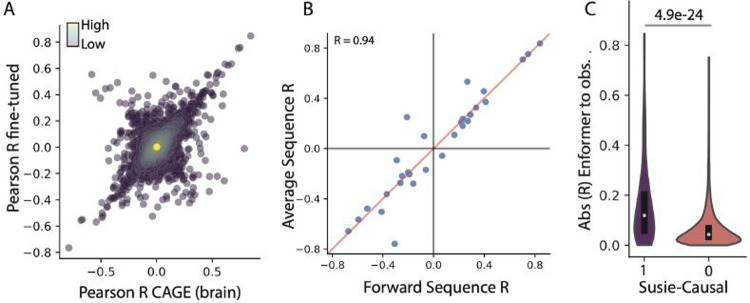
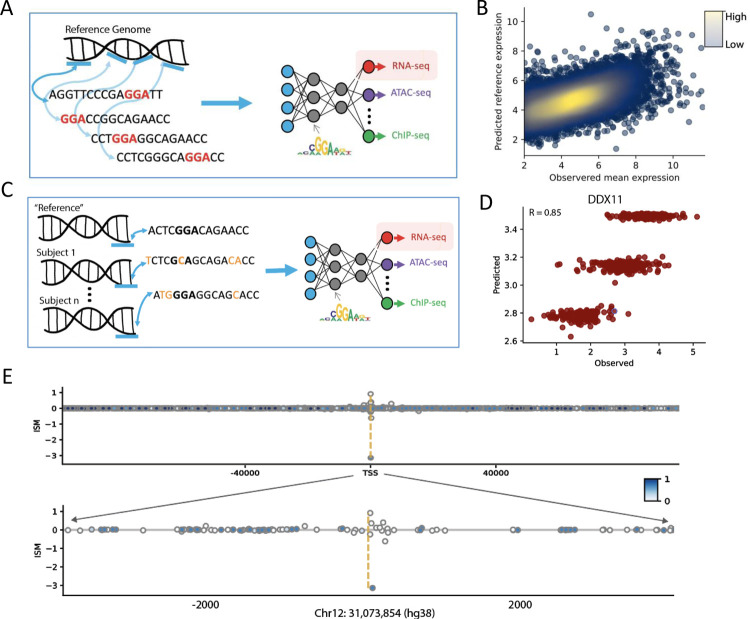
Deep learning methods have recently become the state-of-the-art in a variety of regulatory genomic tasks including the prediction of gene expression from genomic DNA. As such, these methods promise to serve as important tools in interpreting the full spectrum of genetic variation observed in personal genomes. Previous evaluation strategies have assessed their predictions of gene expression across genomic regions, however, systematic benchmarking is lacking to assess their predictions across individuals, which would directly evaluates their utility as personal DNA interpreters. We used paired Whole Genome Sequencing and gene expression from 839 individuals in the ROSMAP study to evaluate the ability of current methods to predict gene expression variation across individuals at varied loci. Our approach identifies a limitation of current methods to correctly predict the direction of variant effects. We show that this limitation stems from insufficiently learnt sequence motif grammar, and suggest new model training strategies to improve performance.
深度学习方法最近已成为包括从基因组DNA预测基因表达在内的各种调控基因组任务中的最先进技术。因此,这些方法有望成为解释在个人基因组中观察到的全谱遗传变异的重要工具。以往的评估策略评估了它们在基因组区域对基因表达的预测,然而,缺乏系统的基准测试来评估它们在个体间的预测,而这将直接评估它们作为个人DNA解释工具的效用。我们利用ROSMAP研究中839名个体的全基因组测序和基因表达配对数据,评估当前方法预测不同位点个体间基因表达变异的能力。我们的方法发现了当前方法在正确预测变异效应方向上的局限性。我们表明,这种局限性源于对序列基序语法学习不足,并提出了新的模型训练策略以提高性能。