Center for Bacterial Pathogenesis, Division of Infectious Diseases, Department of Medicine, Massachusetts General Hospital, Boston, MA 02115.
Department of Microbiology, Blavatnik Institute, Harvard Medical School, Boston, MA 02115.
Proc Natl Acad Sci U S A. 2023 Apr 11;120(15):e2218469120. doi: 10.1073/pnas.2218469120. Epub 2023 Apr 4.
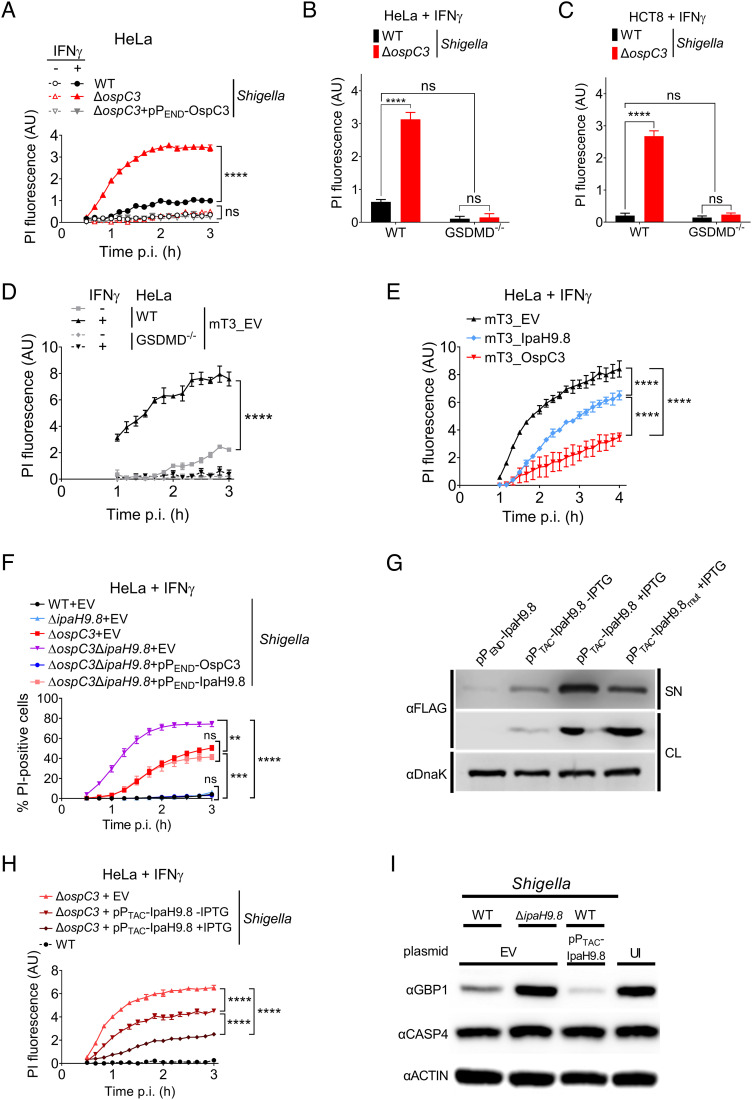
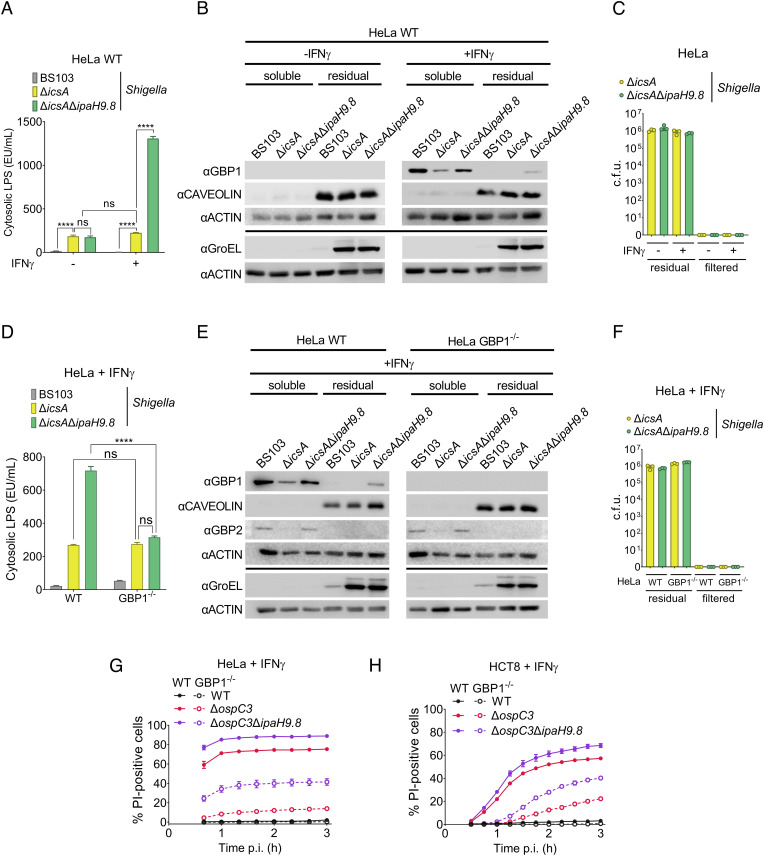
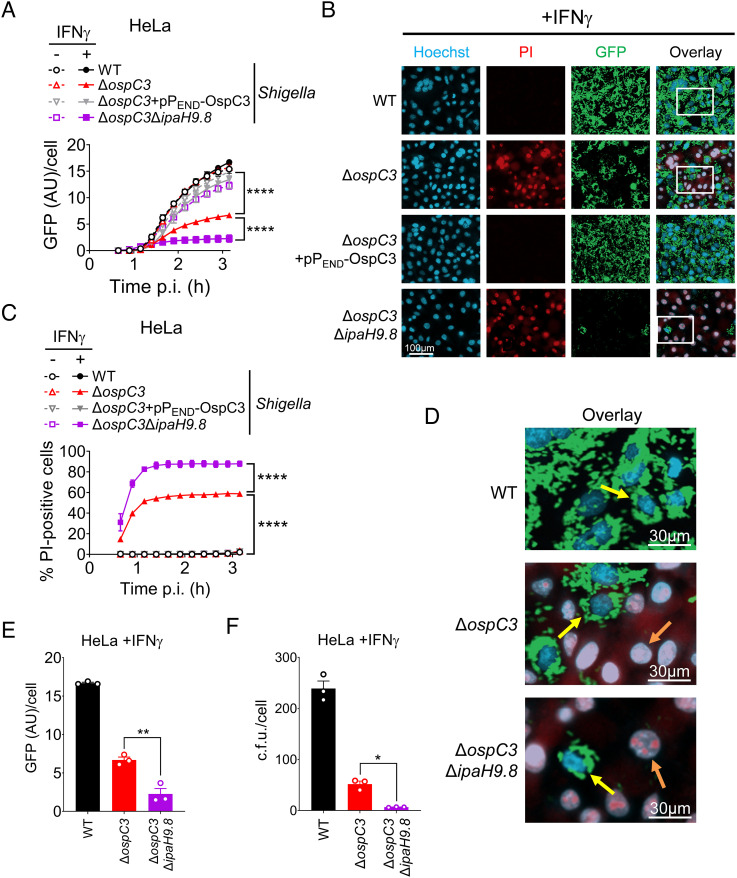
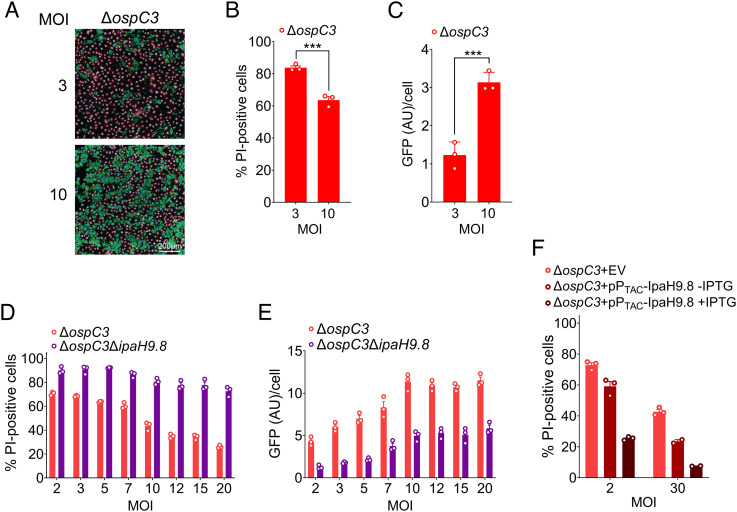
Pyroptosis is an inflammatory form of cell death induced upon recognition of invading microbes. During an infection, pyroptosis is enhanced in interferon-gamma-exposed cells via the actions of members of the guanylate-binding protein (GBP) family. GBPs promote caspase-4 (CASP4) activation by enhancing its interactions with lipopolysaccharide (LPS), a component of the outer envelope of Gram-negative bacteria. Once activated, CASP4 promotes the formation of noncanonical inflammasomes, signaling platforms that mediate pyroptosis. To establish an infection, intracellular bacterial pathogens, like species, inhibit pyroptosis. The pathogenesis of is dependent on its type III secretion system, which injects ~30 effector proteins into host cells. Upon entry into host cells, are encapsulated by GBP1, followed by GBP2, GBP3, GBP4, and in some cases, CASP4. It has been proposed that the recruitment of CASP4 to bacteria leads to its activation. Here, we demonstrate that two effectors, OspC3 and IpaH9.8, cooperate to inhibit CASP4-mediated pyroptosis. We show that in the absence of OspC3, an inhibitor of CASP4, IpaH9.8 inhibits pyroptosis via its known degradation of GBPs. We find that, while some LPS is present within the host cell cytosol of epithelial cells infected with wild-type in the absence of IpaH9.8, increased amounts are shed in a GBP1-dependent manner. Furthermore, we find that additional IpaH9.8 targets, likely GBPs, promote CASP4 activation, even in the absence of GBP1. These observations suggest that by boosting LPS release, GBP1 provides CASP4-enhanced access to cytosolic LPS, thus promoting host cell death via pyroptosis.
细胞焦亡是一种在识别入侵微生物时引发的炎症形式的细胞死亡。在感染过程中,干扰素-γ暴露的细胞中,鸟嘌呤核苷酸结合蛋白(GBP)家族成员的作用增强了细胞焦亡。GBPs 通过增强其与脂多糖(LPS)的相互作用来促进半胱天冬酶-4(CASP4)的激活,LPS 是革兰氏阴性细菌外膜的组成部分。一旦被激活,CASP4 就会促进非典型炎性小体的形成,这是介导细胞焦亡的信号平台。为了建立感染,细胞内细菌病原体,如 ,抑制细胞焦亡。的发病机制依赖于其 III 型分泌系统,该系统将大约 30 种效应蛋白注入宿主细胞。进入宿主细胞后,被 GBP1 包裹,随后是 GBP2、GBP3、GBP4,在某些情况下,还有 CASP4。有人提出,CASP4 被招募到细菌上会导致其激活。在这里,我们证明了两种 效应蛋白,OspC3 和 IpaH9.8,合作抑制 CASP4 介导的细胞焦亡。我们表明,在没有 OspC3(CASP4 的抑制剂)的情况下,IpaH9.8 通过其已知的 GBP 降解来抑制细胞焦亡。我们发现,虽然在没有 IpaH9.8 的情况下,野生型感染的上皮细胞的宿主细胞质中存在一些 LPS,但以 GBP1 依赖的方式释放出更多的 LPS。此外,我们发现,其他 IpaH9.8 靶点,可能是 GBP,即使在没有 GBP1 的情况下,也能促进 CASP4 的激活。这些观察结果表明,通过增加 LPS 的释放,GBP1 为 CASP4 提供了增强的细胞溶质 LPS 进入途径,从而通过细胞焦亡促进宿主细胞死亡。