Center of Clinical Laboratory, Zhongshan Hospital, School of Medicine, Xiamen University, Xiamen, China.
Institute of Infectious Disease, School of Medicine, Xiamen University, Xiamen, China.
Microbiol Spectr. 2023 Jun 15;11(3):e0493122. doi: 10.1128/spectrum.04931-22. Epub 2023 Apr 10.
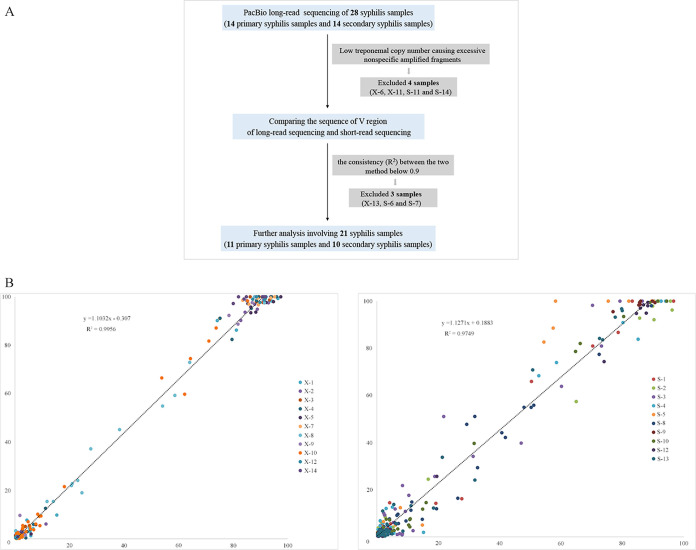
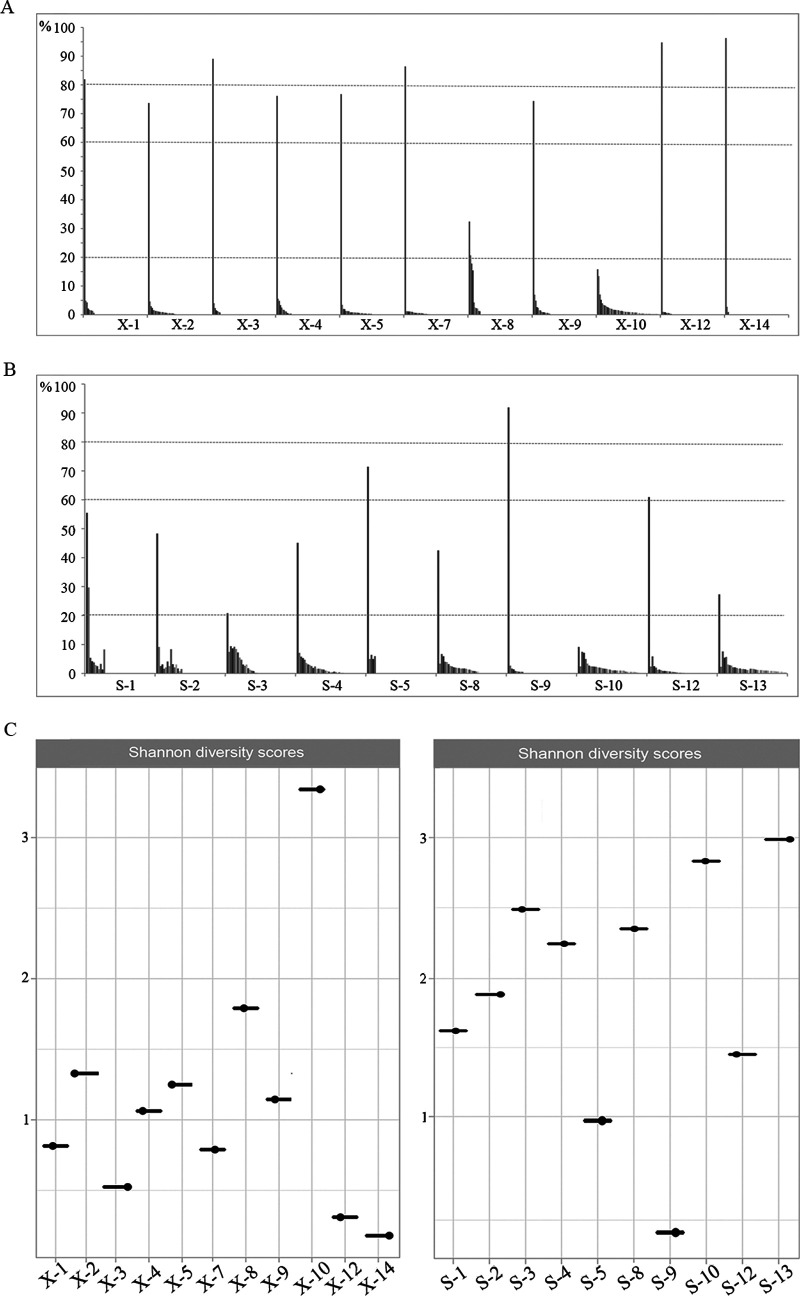
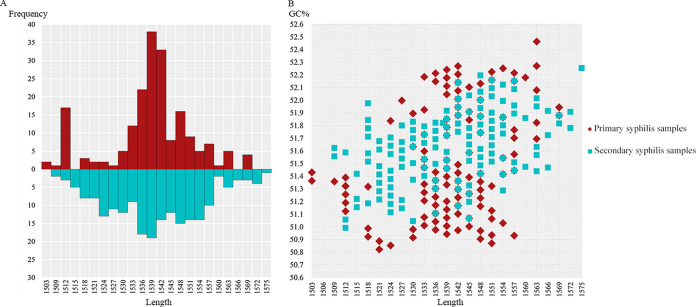
TprK antigenic variation is acknowledged as an important strategy developed by Treponema pallidum to achieve immune evasion. Previous studies applied short-read sequencing to explore gene sequence diversity in clinical samples; however, due to the limitations of short-read sequencing, it was difficult to determine the linkage between the seven V regions, and crucial information about full-length variants was lost. Although two recent studies explored complete gene profiles in natural human syphilis infection, there are still too few profiled full-length variants among clinical T. pallidum isolates to fully understand the characteristics of TprK coding diversity. Here, Pacific Biosciences (PacBio) long-read sequencing was applied to examine the diversity of full-length variants in 21 clinical T. pallidum isolates from 11 patients with primary syphilis and 10 patients with secondary syphilis. A total of 398 high-confidence full-length sequences, which presented remarkable sequence heterogeneity, were found. However, these full-length variants exhibited limited variation in length and GC content, showing 24 length types and average GC content of 51.5 ± 0.42% and 51.6 ± 0.26% for primary and secondary syphilis samples, respectively. Additionally, the combined patterns of mutated V regions generating new variants were obviously different in primary and secondary syphilis samples. The diversity of gene sequences in primary syphilis samples may represent the underlying variability of the bacterium; conversely, the variability of the gene in secondary syphilis samples may more accurately reflect how T. pallidum escapes host immune clearance. These data highlight the gene as an important coding gene that shows conflicting genetic characteristics but underlies the persistence of spirochete infection. The resurgence of syphilis in both low- and high-income countries has attracted attention, and persistent infection by the pathogen has long been a research focus. The gene, encoding the hypervariable outer membrane protein, is thought to be responsible for pathogen immune evasion and persistent infection. Here, PacBio long-read sequencing was applied to examine the diversity of full-length variants in 21 clinical T. pallidum isolates from 11 patients with primary syphilis and 10 patients with secondary syphilis. The results showed that the sequences of the gene were remarkably heterogeneous; however, the sequences presented limited variation in length and GC content. The investigation of the combined patterns of the V regions allowed us to gain insight into the features of the gene generating new variants at different clinical stages. The findings of this study will be helpful for further exploration of the pathogenesis of syphilis.
TprK 抗原变异被认为是梅毒螺旋体逃避免疫的重要策略。先前的研究应用短读长测序来探索临床样本中基因序列的多样性;然而,由于短读长测序的限制,难以确定七个 V 区之间的联系,并且丢失了关于全长 TprK 变异体的重要信息。尽管最近的两项研究探索了自然感染人类梅毒中的完整 TprK 基因谱,但在临床分离的梅毒螺旋体中,仍有太少的全长 TprK 变异体得到充分描述,无法全面了解 TprK 编码多样性的特征。在此,应用太平洋生物科学公司(PacBio)长读长测序来检测 21 例来自 11 例原发性梅毒和 10 例二期梅毒患者的临床梅毒螺旋体分离株中全长 TprK 变异体的多样性。共发现 398 个高可信度的全长序列,它们表现出显著的序列异质性。然而,这些全长 TprK 变异体在长度和 GC 含量上表现出有限的变异,原发性和二期梅毒样本的长度类型分别为 24 种,平均 GC 含量分别为 51.5±0.42%和 51.6±0.26%。此外,在原发性和二期梅毒样本中,产生新变异体的突变 V 区的组合模式明显不同。原发性梅毒样本中 TprK 基因序列的多样性可能代表了细菌的潜在变异性;相反,二期梅毒样本中 TprK 基因的变异性可能更准确地反映了梅毒螺旋体如何逃避宿主免疫清除。这些数据强调了 TprK 基因作为一个重要的编码基因,表现出相互矛盾的遗传特征,但却构成了螺旋体感染的持续性。梅毒在低收入和高收入国家的死灰复燃引起了人们的关注,病原体的持续感染一直是研究的重点。编码高度可变的外膜蛋白的 TprK 基因被认为是导致病原体免疫逃避和持续感染的原因。在此,应用太平洋生物科学公司(PacBio)长读长测序来检测 21 例来自 11 例原发性梅毒和 10 例二期梅毒患者的临床梅毒螺旋体分离株中全长 TprK 变异体的多样性。结果表明,TprK 基因序列高度异质;然而,序列在长度和 GC 含量上表现出有限的变异。对 V 区组合模式的研究使我们能够深入了解在不同临床阶段产生新变异体的 TprK 基因的特征。本研究的发现将有助于进一步探索梅毒的发病机制。