Division of Cardiology, Department of Medicine, Duke University Medical Center, Durham, North Carolina, USA.
Department of Biochemistry and Molecular Medicine, University of Montreal, Montreal, Quebec, Canada; Institute for Research in Immunology and Cancer, University of Montreal, Montreal, Quebec, Canada.
J Biol Chem. 2023 May;299(5):104690. doi: 10.1016/j.jbc.2023.104690. Epub 2023 Apr 8.
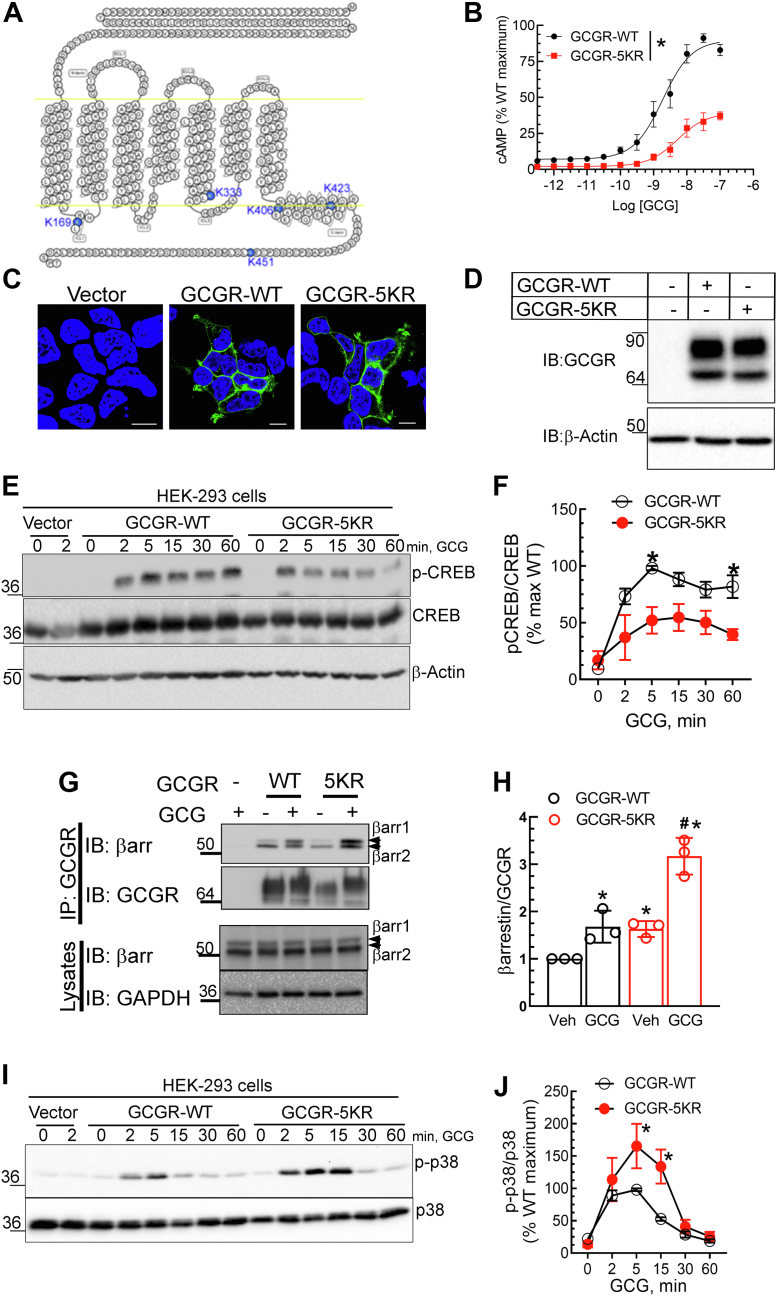
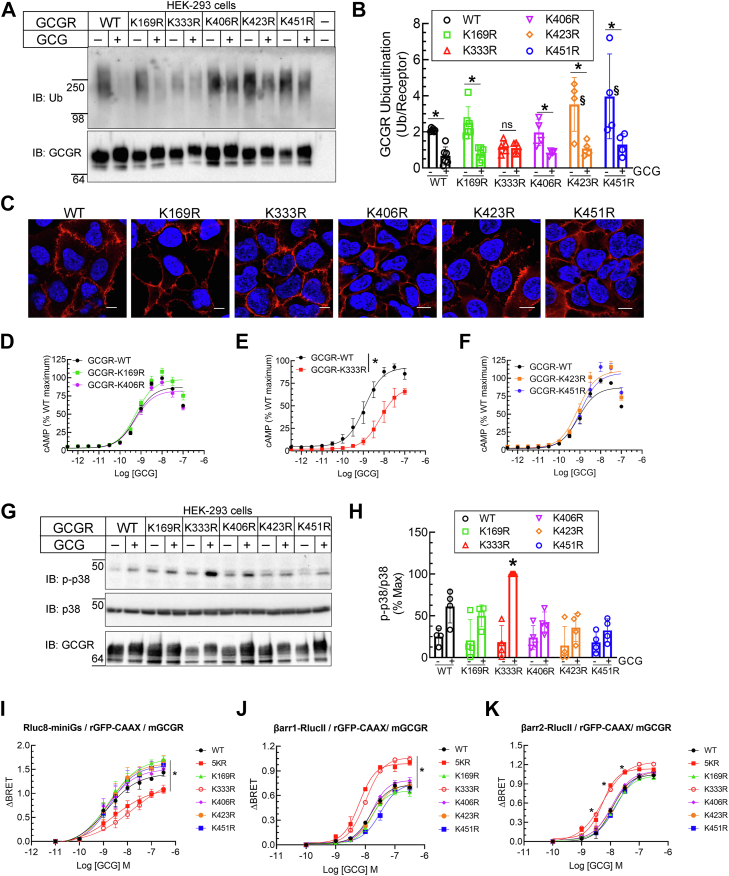
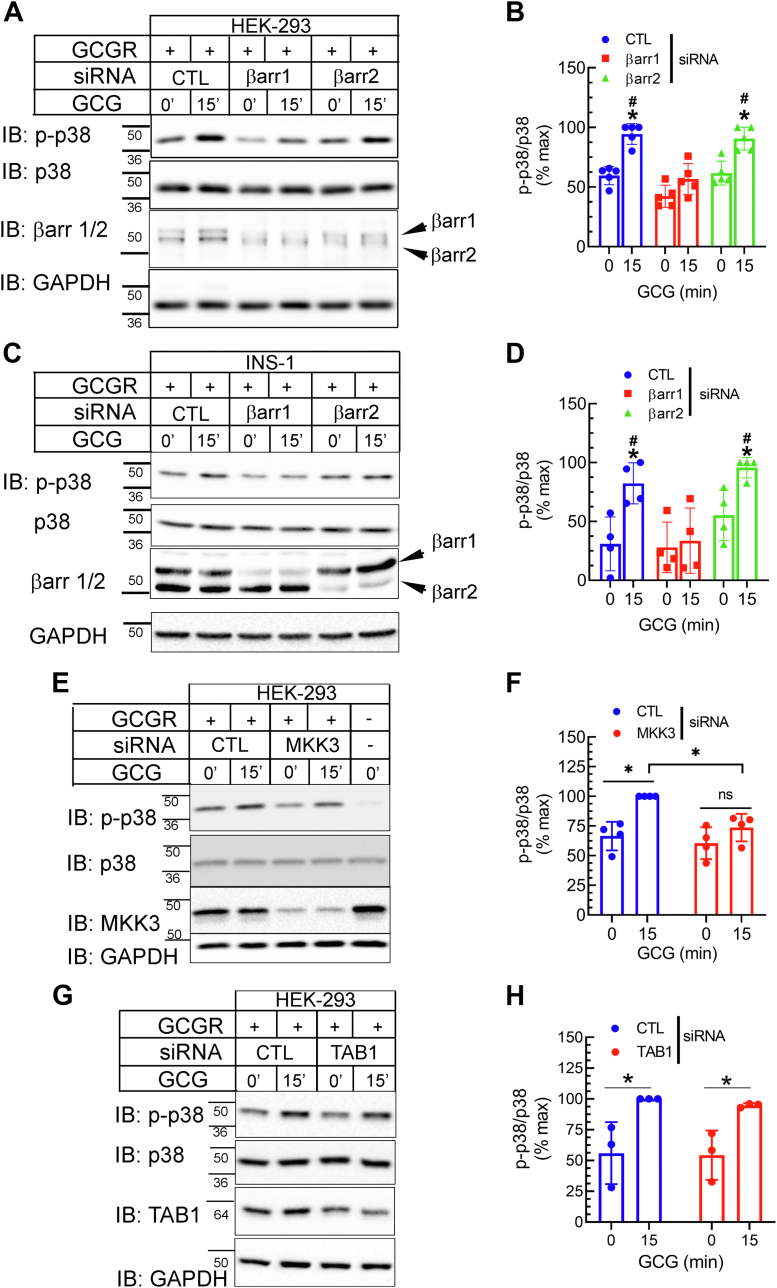
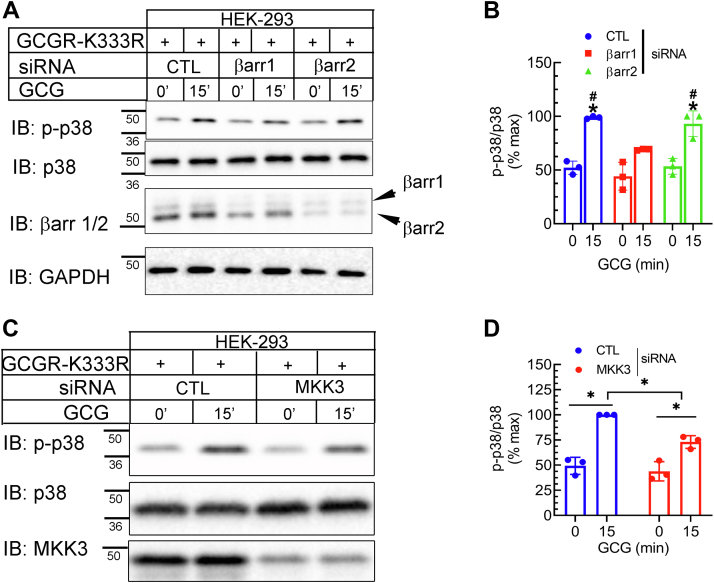
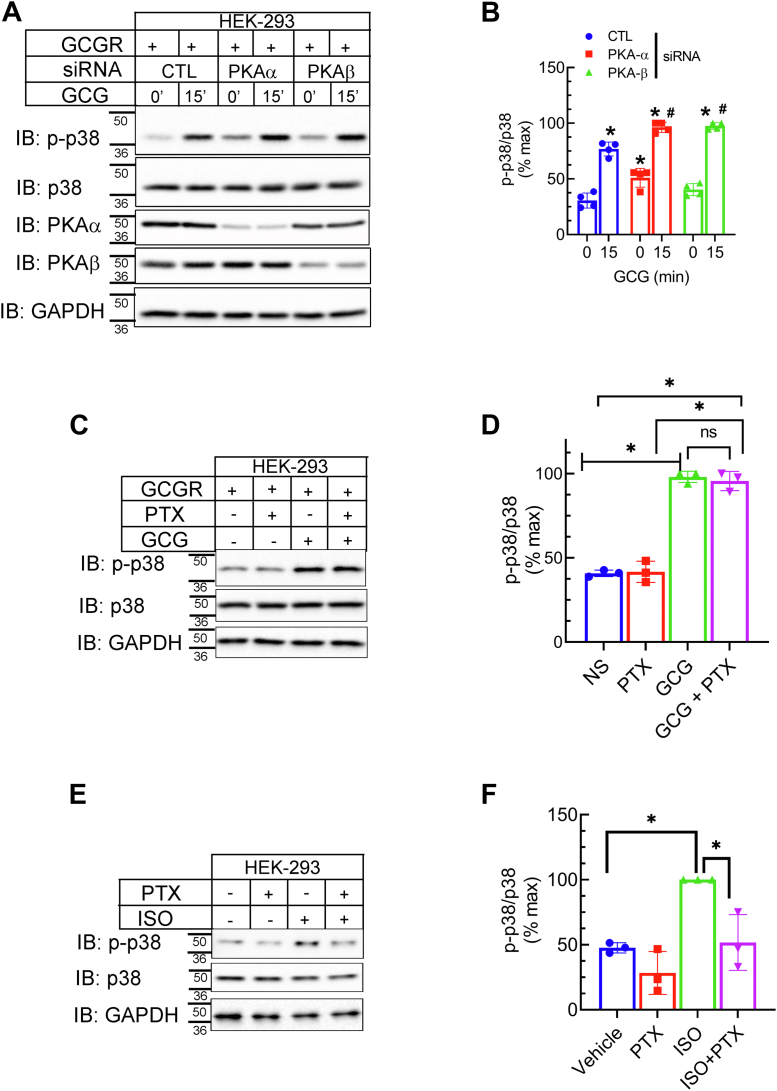
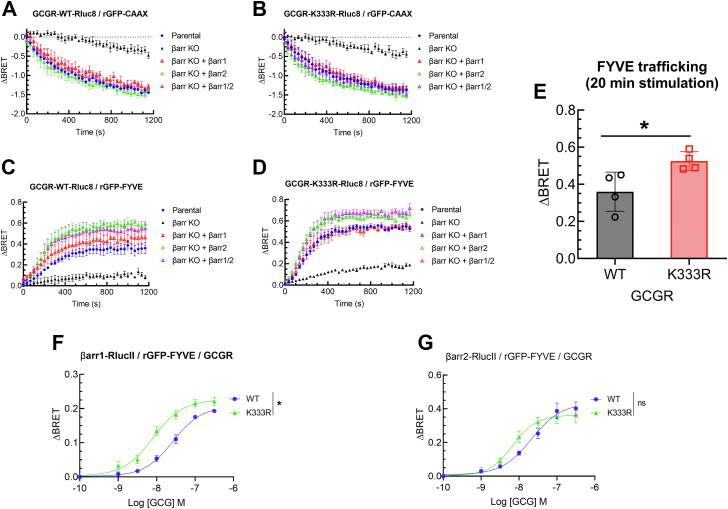
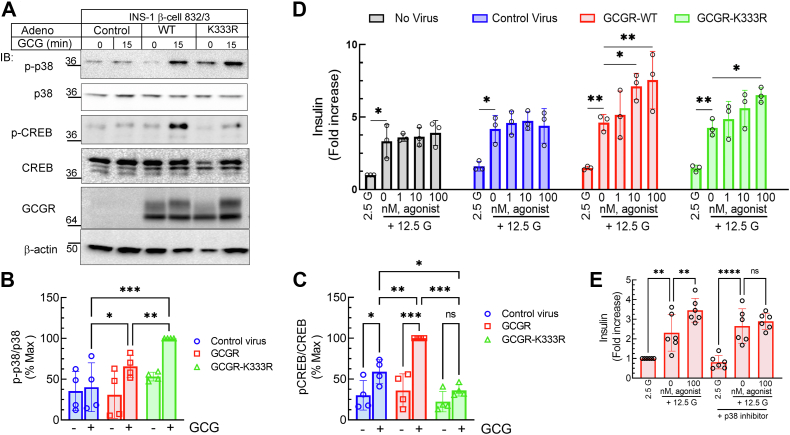
The pancreatic hormone glucagon activates the glucagon receptor (GCGR), a class B seven-transmembrane G protein-coupled receptor that couples to the stimulatory heterotrimeric G protein and provokes PKA-dependent signaling cascades vital to hepatic glucose metabolism and islet insulin secretion. Glucagon-stimulation also initiates recruitment of the endocytic adaptors, βarrestin1 and βarrestin2, which regulate desensitization and internalization of the GCGR. Unlike many other G protein-coupled receptors, the GCGR expressed at the plasma membrane is constitutively ubiquitinated and upon agonist-activation, internalized GCGRs are deubiquitinated at early endosomes and recycled via Rab4-containing vesicles. Herein we report a novel link between the ubiquitination status and signal transduction mechanism of the GCGR. In the deubiquitinated state, coupling of the GCGR to Gs is diminished, while binding to βarrestin is enhanced with signaling biased to a βarrestin1-dependent p38 mitogen activated protein kinase (MAPK) pathway. This ubiquitin-dependent signaling bias arises through the modification of lysine333 (K333) on the cytoplasmic face of transmembrane helix V. Compared with the GCGR-WT, the mutant GCGR-K333R has impaired ubiquitination, diminished G protein coupling, and PKA signaling but unimpaired potentiation of glucose-stimulated-insulin secretion in response to agonist-stimulation, which involves p38 MAPK signaling. Both WT and GCGR-K333R promote the formation of glucagon-induced βarrestin1-dependent p38 signaling scaffold that requires canonical upstream MAPK-Kinase3, but is independent of Gs, Gi, and βarrestin2. Thus, ubiquitination/deubiquitination at K333 in the GCGR defines the activation of distinct transducers with the potential to influence various facets of glucagon signaling in health and disease.
胰腺激素胰高血糖素激活胰高血糖素受体(GCGR),这是一种 B 类七跨膜 G 蛋白偶联受体,与刺激性异三聚体 G 蛋白偶联,并引发 PKA 依赖性信号级联反应,对肝葡萄糖代谢和胰岛胰岛素分泌至关重要。胰高血糖素刺激还启动了内吞衔接蛋白βarrestin1 和 βarrestin2 的募集,它们调节 GCGR 的脱敏和内化。与许多其他 G 蛋白偶联受体不同,在质膜上表达的 GCGR 是组成型泛素化的,并且在激动剂激活后,内化的 GCGR 在早期内体中去泛素化,并通过含有 Rab4 的囊泡回收。本文报道了 GCGR 的泛素化状态和信号转导机制之间的新联系。在去泛素化状态下,GCGR 与 Gs 的偶联减少,而与βarrestin 的结合增强,信号偏向于βarrestin1 依赖性 p38 有丝分裂原激活蛋白激酶(MAPK)途径。这种依赖于泛素的信号偏向是通过跨膜螺旋 V 的细胞质面赖氨酸 333(K333)的修饰产生的。与 GCGR-WT 相比,突变 GCGR-K333R 泛素化受损,G 蛋白偶联减少,PKA 信号减弱,但对激动剂刺激下葡萄糖刺激的胰岛素分泌的增强作用不变,这涉及到 p38 MAPK 信号。WT 和 GCGR-K333R 均促进形成胰高血糖素诱导的βarrestin1 依赖性 p38 信号支架,该支架需要经典的上游 MAPK-激酶 3,但不依赖于 Gs、Gi 和βarrestin2。因此,GCGR 中的 K333 泛素化/去泛素化定义了不同转导器的激活,有可能影响健康和疾病中胰高血糖素信号的各个方面。