Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, United States.
The Institute for Experiential AI, Northeastern University, Boston, Massachusetts 02115, United States.
J Chem Theory Comput. 2023 May 9;19(9):2469-2483. doi: 10.1021/acs.jctc.2c01128. Epub 2023 Apr 11.
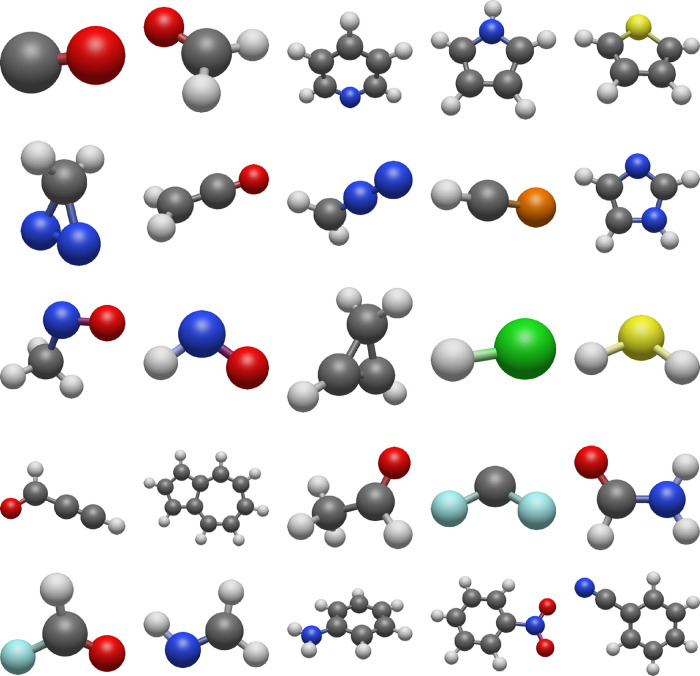
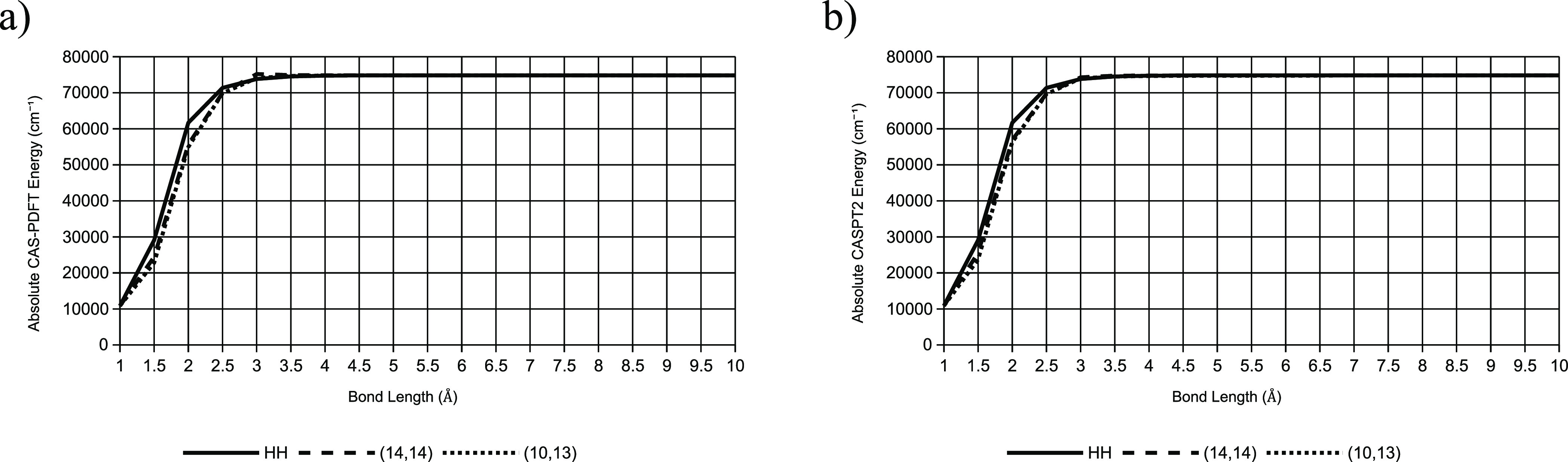
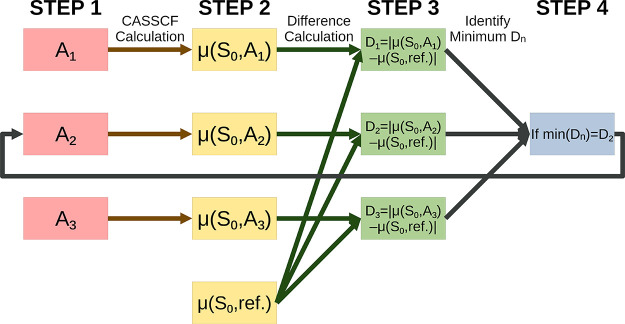
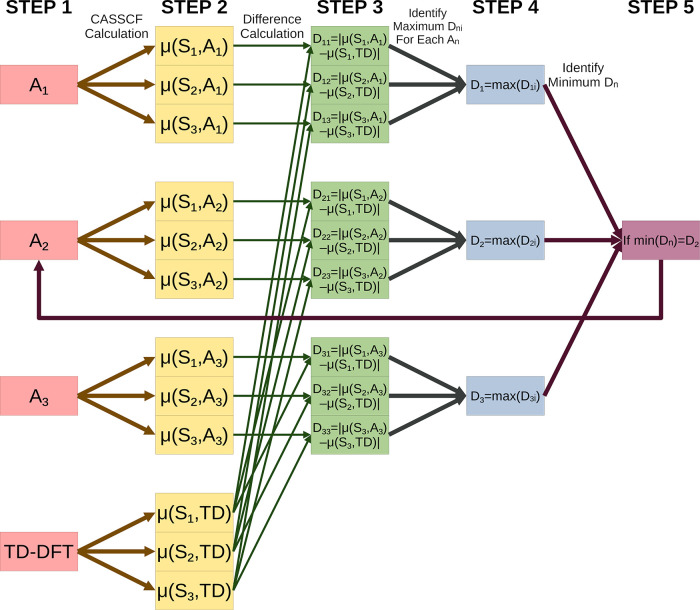
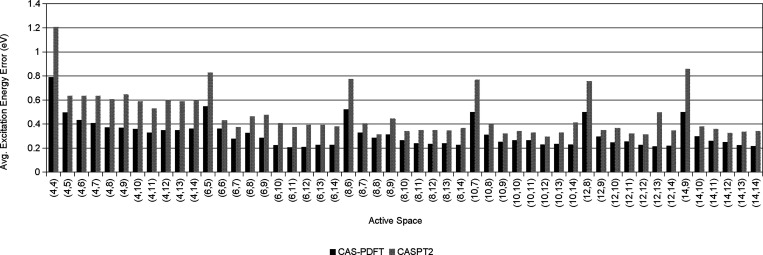
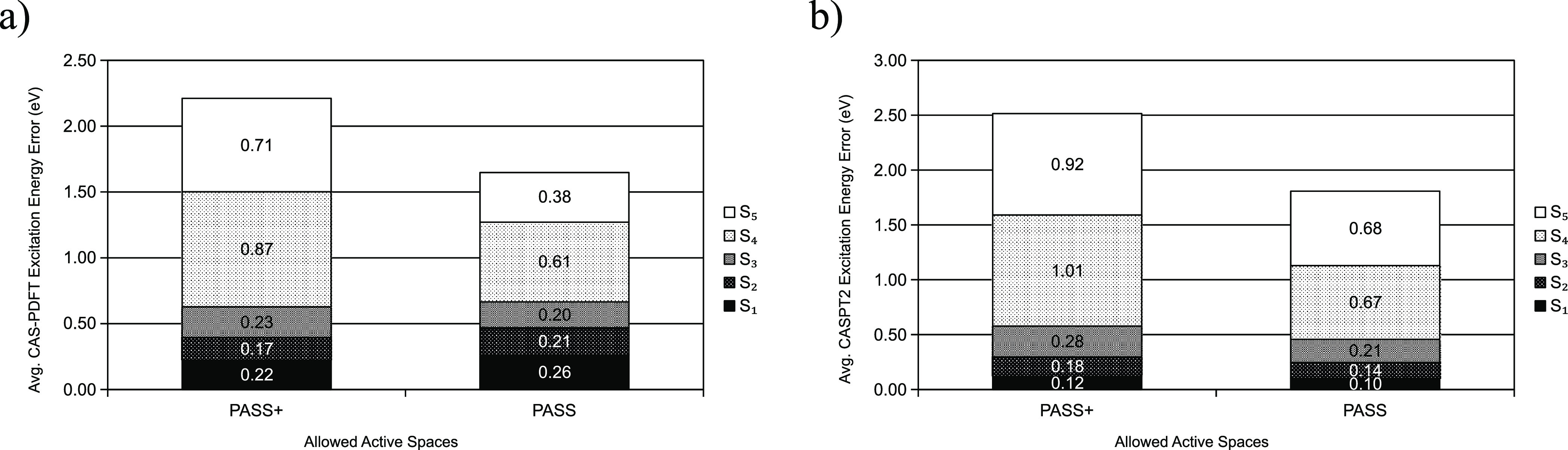
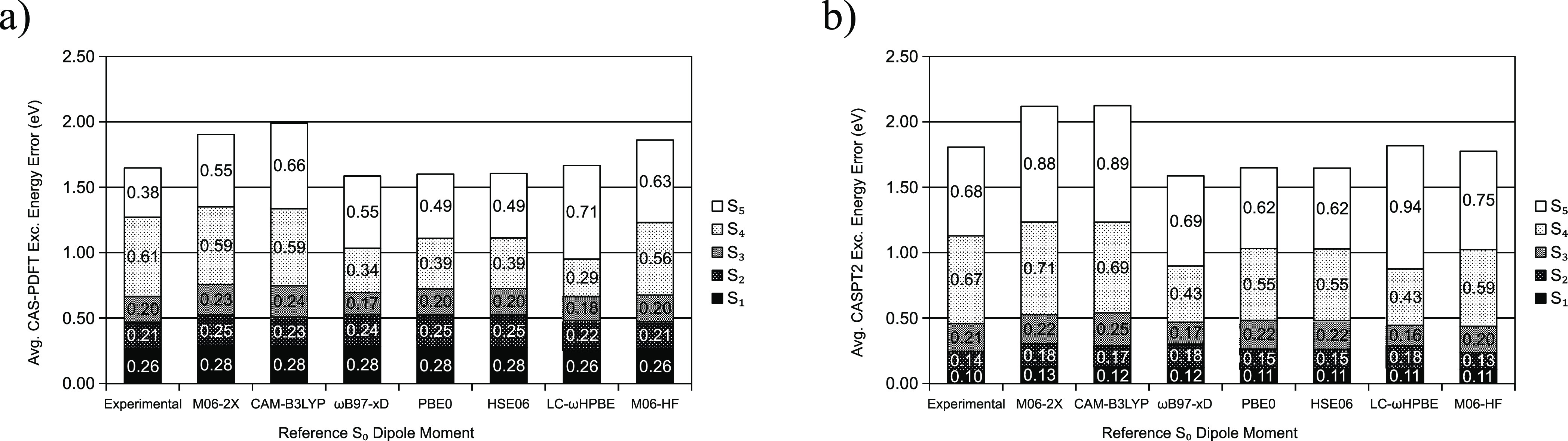
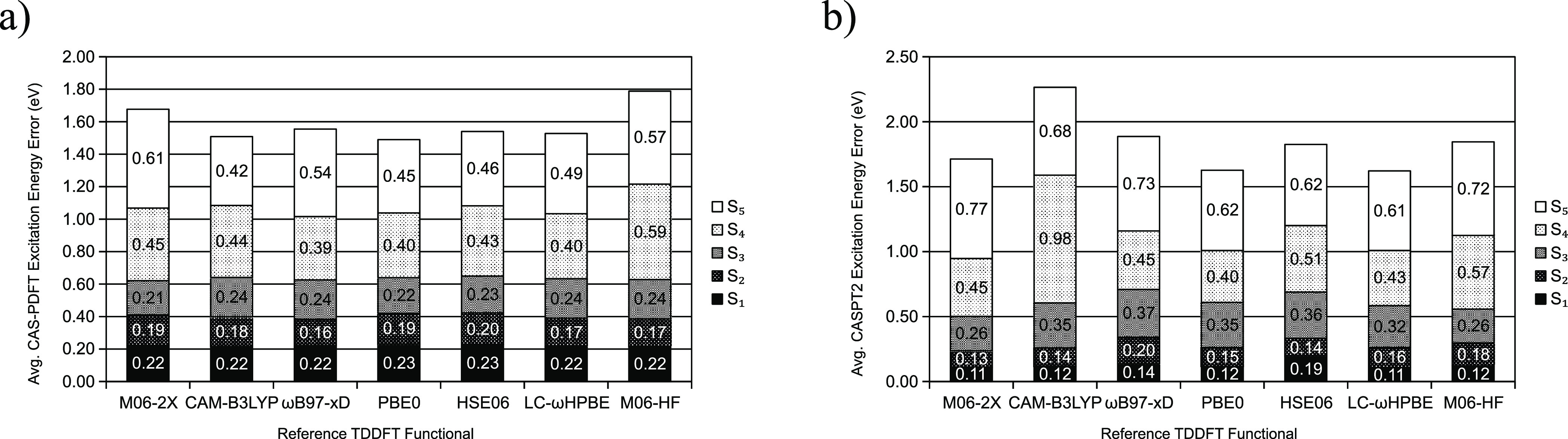
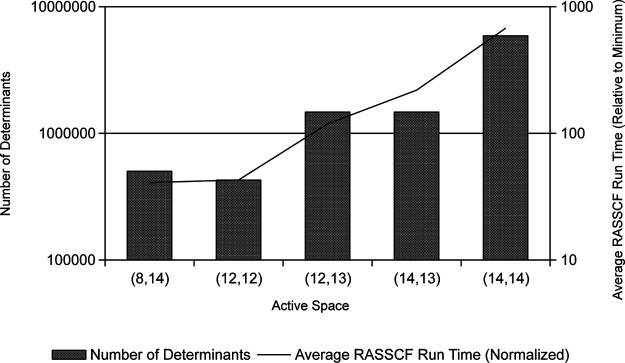
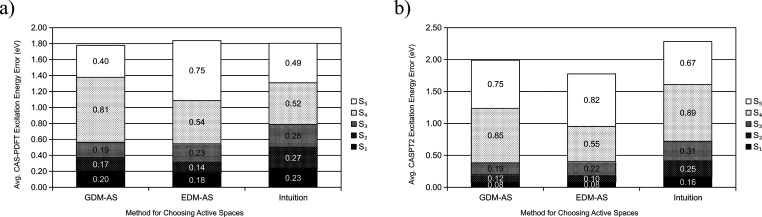
Multireference calculations can provide accurate information of systems with strong correlation, which have increasing importance in the development of new molecules and materials. However, selecting a suitable active space for multireference calculations is nontrivial, and the selection of an unsuitable active space can sometimes lead to results that are not physically meaningful. Active space selection often requires significant human input, and the selection that leads to reasonable results often goes beyond chemical intuition. In this work, we have developed and evaluated two protocols for automated selection of the active space for multireference calculations based on a simple physical observable, the dipole moment, for molecules with nonzero ground-state dipole moments. One protocol is based on the ground-state dipole moment, and the other is based on the excited-state dipole moments. To evaluate the protocols, we constructed a dataset of 1275 active spaces from 25 molecules, each with 51 active space sizes considered, and have mapped out the relationship between the active space, dipole moments, and vertical excitation energies. We have demonstrated that, within this dataset, our protocols allow one to choose among a number of accessible active spaces one that is likely to give reasonable vertical excitation energies, especially for the first three excitations, with no parameters manually decided by the user. We show that, with large active spaces removed from consideration, the accuracy is similar and the time-to-solution can be reduced by more than 10 fold. We also show that the protocols can be applied to potential energy surface scans and determining the spin states of transition metal oxides.
多参考计算可以为强相关系统提供准确的信息,这在新分子和材料的开发中越来越重要。然而,选择合适的多参考计算活动空间并不简单,选择不合适的活动空间有时会导致没有物理意义的结果。活动空间选择通常需要大量的人工输入,而导致合理结果的选择往往超出了化学直觉。在这项工作中,我们开发并评估了两种基于简单物理量(分子的基态偶极矩)的自动选择多参考计算活动空间的协议,对于具有非零基态偶极矩的分子。一个协议基于基态偶极矩,另一个协议基于激发态偶极矩。为了评估协议,我们构建了一个包含 1275 个活性空间的数据集,这些活性空间来自 25 个分子,每个分子考虑了 51 种活性空间大小,并绘制了活性空间、偶极矩和垂直激发能之间的关系。我们已经证明,在这个数据集中,我们的协议可以在许多可访问的活性空间中进行选择,选择一个可能给出合理垂直激发能的活性空间,特别是对于前三个激发态,不需要用户手动决定任何参数。我们表明,在去除较大的活动空间后,准确性相似,求解时间可以减少 10 倍以上。我们还表明,该协议可应用于势能面扫描和确定过渡金属氧化物的自旋态。