Kabir Mohammad Pabel, Ghosh Paulami, Gozem Samer
Department of Chemistry, Georgia State University, Atlanta, Georgia 30302, United States.
J Phys Chem B. 2024 Aug 8;128(31):7545-7557. doi: 10.1021/acs.jpcb.4c03748. Epub 2024 Jul 29.
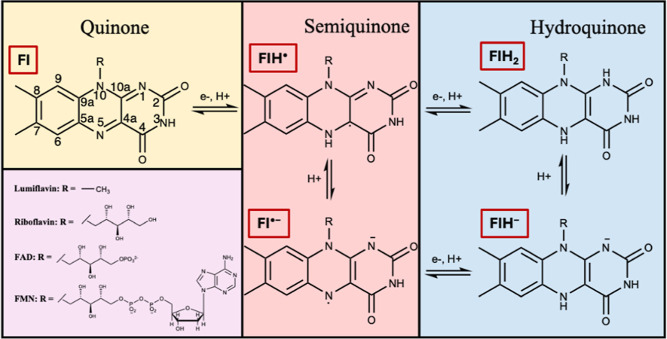
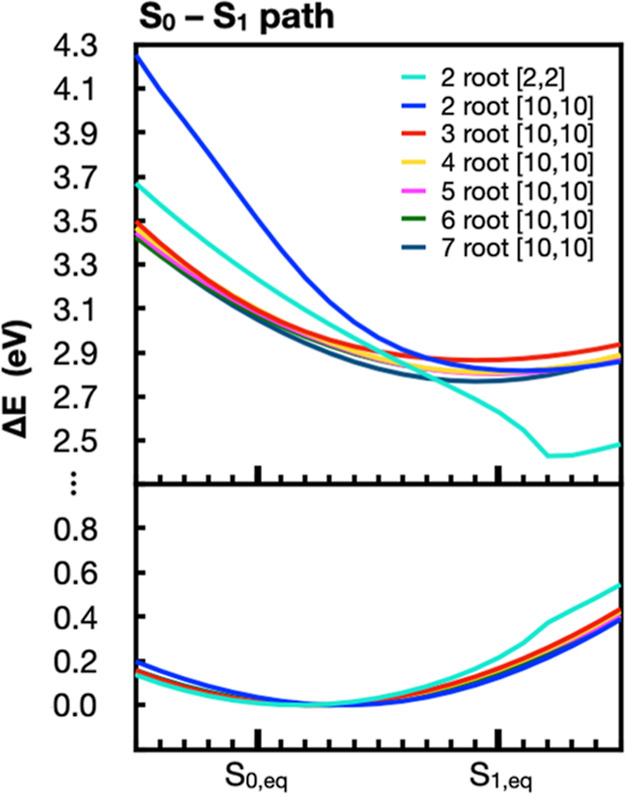
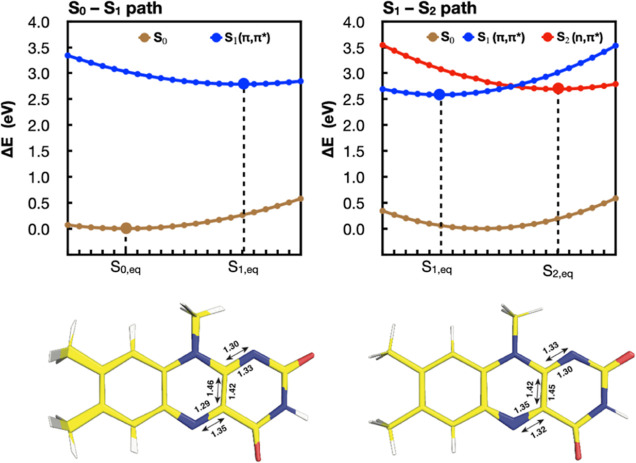
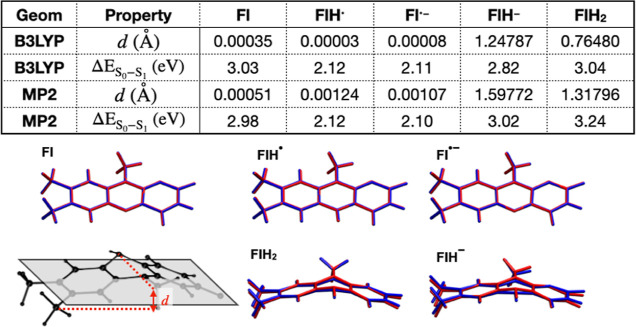
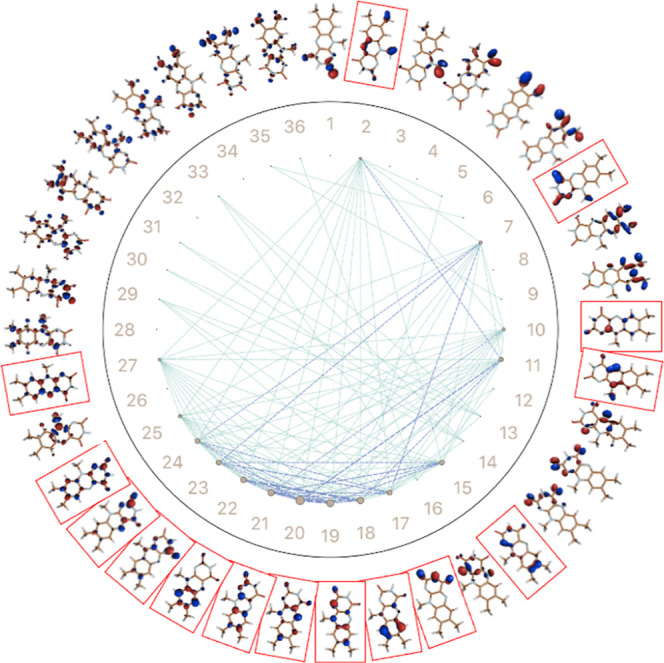
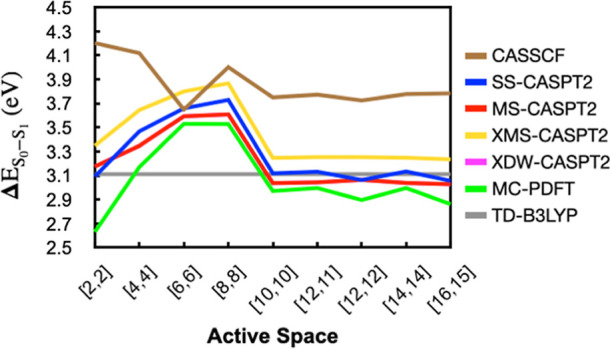
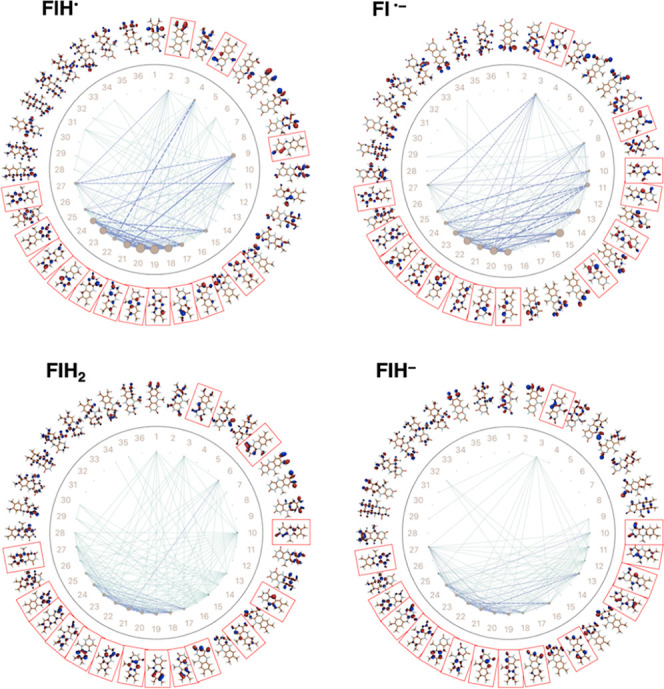
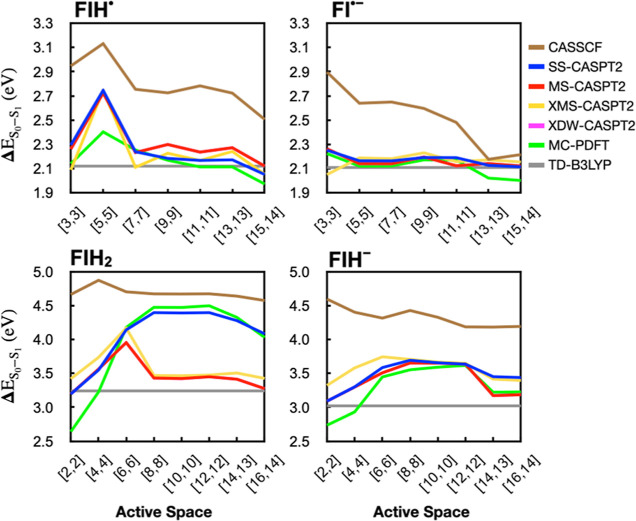
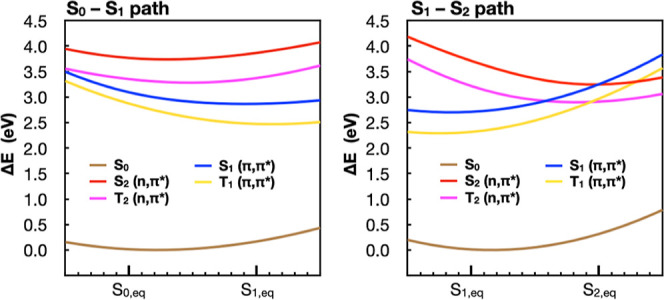
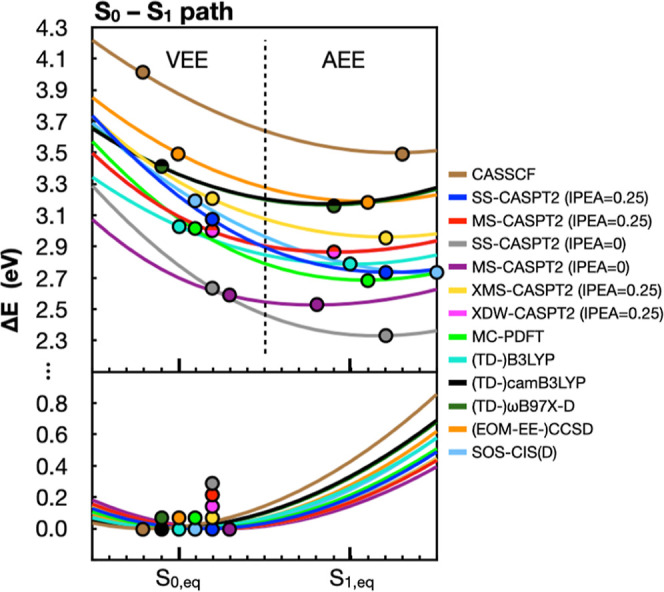
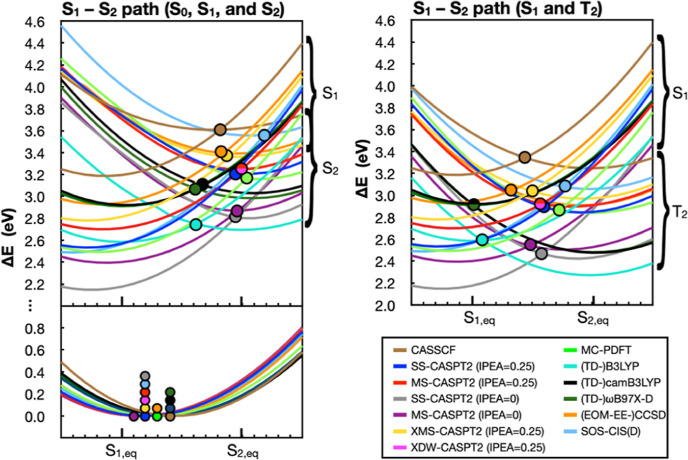
The use of flavins and flavoproteins in photocatalytic, sensing, and biotechnological applications has led to a growing interest in computationally modeling the excited-state electronic structure and photophysics of flavin. However, there is limited consensus regarding which computational methods are appropriate for modeling flavin's photophysics. We compare the energies of low-lying excited states of flavin computed with time-dependent density functional theory (TD-DFT), equation-of-motion coupled cluster (EOM-EE-CCSD), scaled opposite-spin configuration interaction [SOS-CIS(D)], multiconfiguration pair-density functional theory (MC-PDFT), and several multireference perturbation theory (MR-PT2) methods. In the first part, we focus on excitation energies of the first singlet excited state (S) of five different redox and protonation states of flavin, with the goal of finding a suitable active space for MR-PT2 calculations. In the second part, we construct two sets of one-dimensional potential energy surfaces connecting the S and S equilibrium geometries (S-S path) and the S (π,π*) and S (,π*) equilibrium geometries (S-S path). The first path therefore follows a Franck-Condon active mode of flavin while the second path maps crossings points between low-lying singlet and triplet states in flavin. We discuss the similarities and differences in the TD-DFT, EOM-EE-CCSD, SOS-CIS(D), MC-PDFT and MR-PT2 energy profiles along these paths. We find that (TD-)DFT methods are suitable for applications such as simulating the spectra of flavins but are inconsistent with several other methods when used for some geometry optimizations and when describing the energetics of dark (,π*) states. MR-PT2 methods show promise for the simulation of flavin's low-lying excited states, but the selection of orbitals for the active space and the number of roots used for state averaging must be done carefully to avoid artifacts. Some properties, such as the intersystem crossing geometry and energy between the S (π,π*) and T (,π*) states, may require additional benchmarking before they can be determined quantitatively.
黄素和黄素蛋白在光催化、传感及生物技术应用中的使用,引发了人们对通过计算模拟黄素激发态电子结构和光物理性质的日益浓厚兴趣。然而,对于哪种计算方法适用于模拟黄素的光物理性质,目前尚未达成广泛共识。我们比较了用含时密度泛函理论(TD-DFT)、运动方程耦合簇方法(EOM-EE-CCSD)、缩放反对称自旋组态相互作用方法[SOS-CIS(D)]、多组态对密度泛函理论(MC-PDFT)以及几种多参考微扰理论(MR-PT2)方法计算得到的黄素低激发态能量。在第一部分,我们着重研究黄素五种不同氧化还原和质子化状态下第一单重激发态(S)的激发能,目的是为MR-PT2计算找到合适的活性空间。在第二部分,我们构建了两组一维势能面,分别连接S和S平衡几何构型(S-S路径)以及S(π,π*)和S(,π*)平衡几何构型(S-S路径)。因此,第一条路径遵循黄素的弗兰克-康登活性模式,而第二条路径描绘了黄素中低单重态和三重态之间的交叉点。我们讨论了沿这些路径TD-DFT、EOM-EE-CCSD、SOS-CIS(D)、MC-PDFT和MR-PT2能量分布的异同。我们发现,(TD-)DFT方法适用于模拟黄素光谱等应用,但在用于某些几何结构优化以及描述暗态(,π*)能量时,与其他几种方法不一致。MR-PT2方法在模拟黄素低激发态方面显示出前景,但必须谨慎选择活性空间的轨道以及用于态平均的根的数量,以避免出现假象。一些性质,如S(π,π*)和T(,π*)态之间的系间窜越几何结构和能量,在能够进行定量确定之前,可能需要额外的基准测试。