Department of Pediatrics, Takatsuki General Hospital, Takatsuki, Osaka, Japan.
Department of Pediatrics, Japanese Red Cross Kyoto Daiichi Hospital, Kyoto, Japan.
Am J Case Rep. 2023 Apr 13;24:e938396. doi: 10.12659/AJCR.938396.
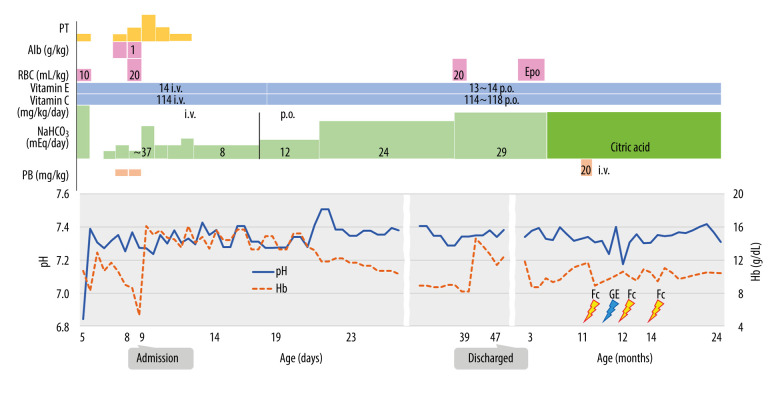
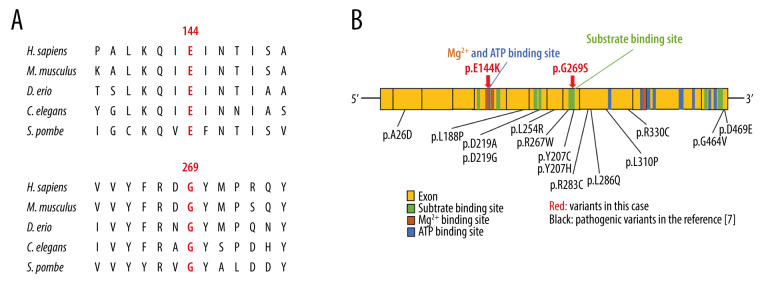
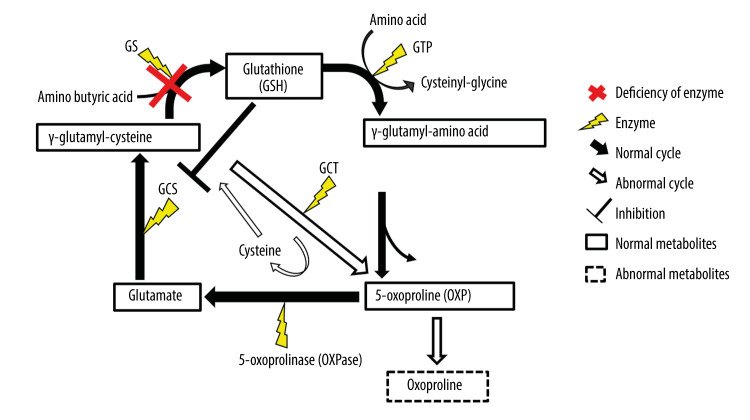
BACKGROUND Glutathione synthetase deficiency (GSD) is a rare autosomal recessive disorder caused by glutathione synthetase (GSS) gene variants that occur in 1 in 1 million individuals. The severe form of GSD is characterized by hemolytic anemia, metabolic acidosis with 5-oxoprolinuria, progressive neurological symptoms, and recurrent bacterial infections. This case report presents a male Japanese infant with severe hemolytic anemia and metabolic acidosis at birth caused by GSD, who developed progressive neurological symptoms on follow-up. CASE REPORT A Japanese male term infant developed severe hemolytic anemia and metabolic acidosis in the early neonatal period. We suspected GSD based on his symptoms and a high 5-oxoproline urine concentration. We began correcting his metabolic acidosis and administering vitamins C and E supplements. The patient required blood transfusion twice during the acute phase for hemolytic anemia. After age 1 month, he maintained good control of metabolic acidosis and hemolytic anemia. A definitive diagnosis of GSD was made based on high concentrations of 5-oxoproline in urine, low concentrations of glutathione and GSS activity in erythrocytes, and genetic testing. Several episodes of febrile convulsions were started at age 11 months, but none occurred after 2 years. At the last follow-up at age 25 months, metabolic acidosis and hemolytic anemia were well controlled, but he had mild neurodevelopmental delay. CONCLUSIONS This case report shows that GSD can present with severe hemolytic anemia and metabolic acidosis at birth, and manifest with subsequent neurological impairment despite early diagnosis and treatment. Therefore, a careful long-term follow-up that includes neurological evaluation is essential for patients with GSD.
谷胱甘肽合酶缺乏症(GSD)是一种罕见的常染色体隐性遗传病,由谷胱甘肽合酶(GSS)基因突变引起,发病率为 1/100 万。严重型 GSD 的特征为溶血性贫血、代谢性酸中毒伴 5-氧脯氨酸尿、进行性神经症状和反复细菌感染。本病例报告介绍了一名日本男性婴儿,出生时因 GSD 出现严重溶血性贫血和代谢性酸中毒,随后出现进行性神经症状。
一名足月出生的日本男性婴儿,在新生儿早期出现严重溶血性贫血和代谢性酸中毒。根据其症状和 5-氧脯氨酸尿浓度升高,我们怀疑为 GSD。我们开始纠正代谢性酸中毒和补充维生素 C 和 E。在急性期,患儿因溶血性贫血接受了 2 次输血。1 月龄后,患儿代谢性酸中毒和溶血性贫血得到良好控制。根据尿中 5-氧脯氨酸浓度升高、红细胞中谷胱甘肽和 GSS 活性降低以及基因检测结果,最终确诊为 GSD。11 月龄时患儿开始出现数次热性惊厥,但 2 岁后未再发作。在 25 月龄的最后一次随访中,代谢性酸中毒和溶血性贫血得到良好控制,但存在轻度神经发育迟缓。
本病例报告表明,GSD 可在出生时表现为严重溶血性贫血和代谢性酸中毒,并在早期诊断和治疗后出现进行性神经损伤。因此,对于 GSD 患者,需要进行仔细的长期随访,包括神经评估。