Department of Ecology and Evolutionary Biology, University of California, Los Angeles, California 90095-1606, USA; Department of Human Genetics, David Geffen School of Medicine, University of California, Los Angeles, California 90095, USA
Genome Res. 2023 Apr;33(4):632-643. doi: 10.1101/gr.276386.121. Epub 2023 Apr 13.
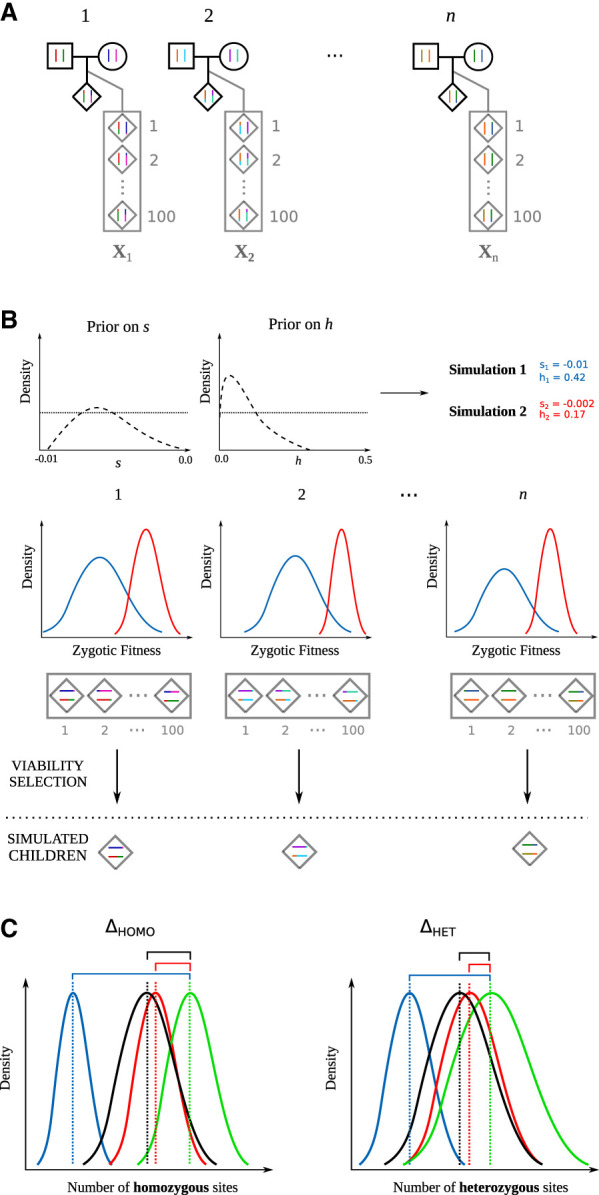
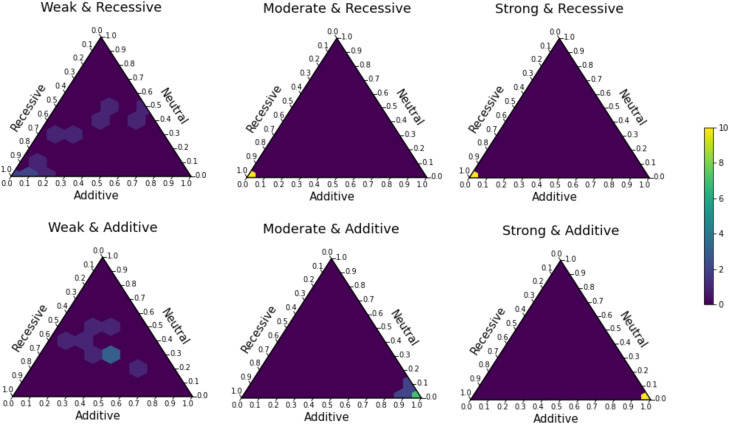
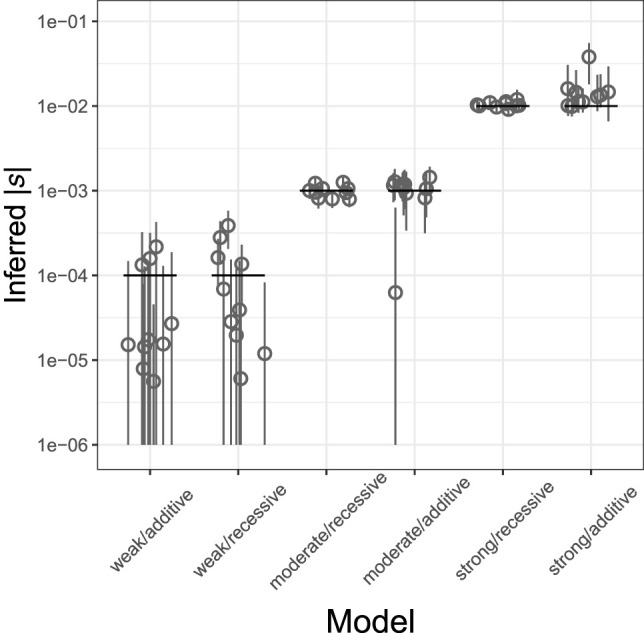
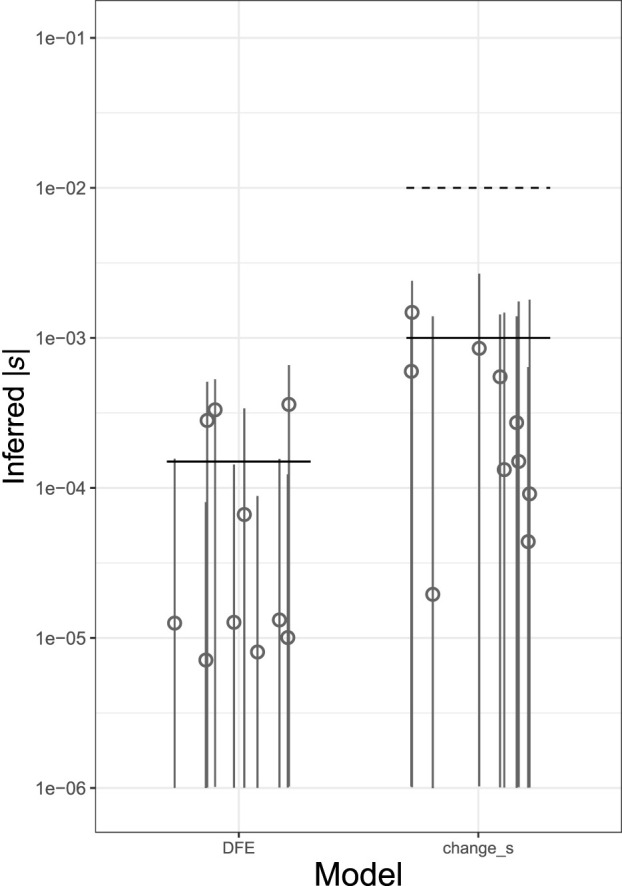
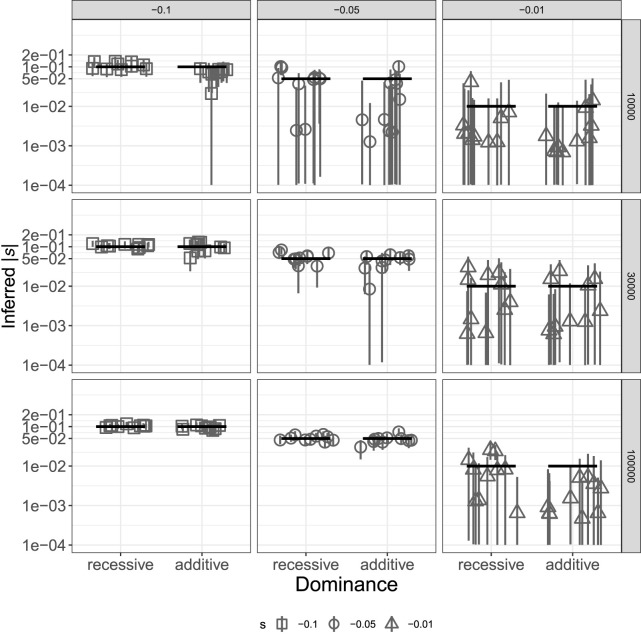
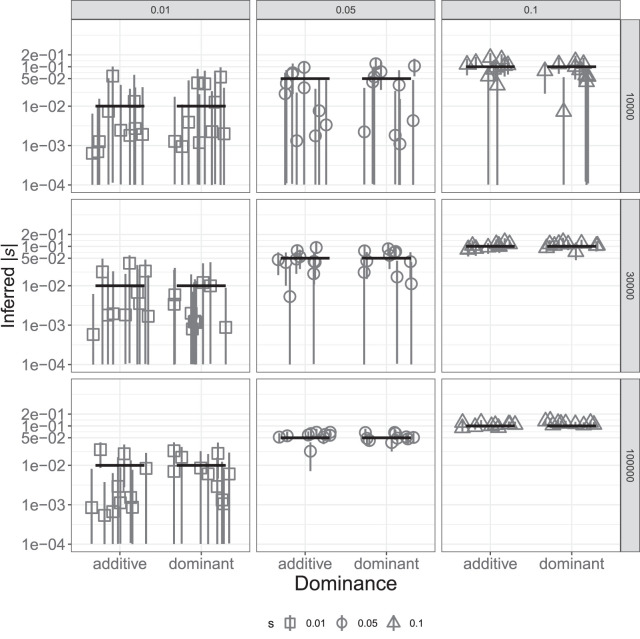
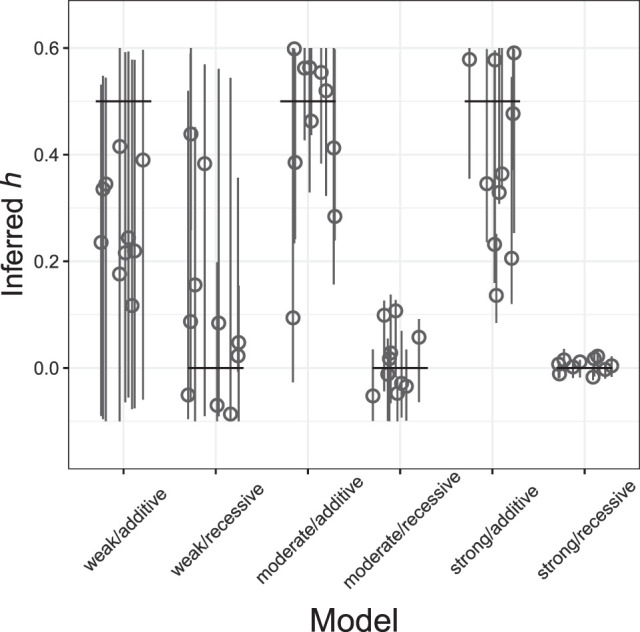
Genome sequence data are no longer scarce. The UK Biobank alone comprises 200,000 individual genomes, with more on the way, leading the field of human genetics toward sequencing entire populations. Within the next decades, other model organisms will follow suit, especially domesticated species such as crops and livestock. Having sequences from most individuals in a population will present new challenges for using these data to improve health and agriculture in the pursuit of a sustainable future. Existing population genetic methods are designed to model hundreds of randomly sampled sequences but are not optimized for extracting the information contained in the larger and richer data sets that are beginning to emerge, with thousands of closely related individuals. Here we develop a new method called trio-based inference of dominance and selection (TIDES) that uses data from tens of thousands of family trios to make inferences about natural selection acting in a single generation. TIDES further improves on the state of the art by making no assumptions regarding demography, linkage, or dominance. We discuss how our method paves the way for studying natural selection from new angles.
基因组序列数据不再稀缺。仅英国生物银行就包含了 20 万个个体基因组,而且还会有更多的基因组数据,这使得人类遗传学领域朝着对整个人群进行测序的方向发展。在未来几十年内,其他模式生物也将效仿,尤其是像农作物和家畜这样的驯化物种。拥有一个种群中大多数个体的序列将为利用这些数据来改善健康和农业,以追求可持续的未来带来新的挑战。现有的群体遗传学方法旨在对数百个随机抽样的序列进行建模,但对于从更大、更丰富的数据集(其中包含数千个密切相关的个体)中提取信息的能力并不是最优的。在这里,我们开发了一种新的方法,称为基于三亲体的显性和选择推断(TIDES),该方法使用来自数万个家族三亲体的数据,对在单一代中起作用的自然选择进行推断。TIDES 进一步改进了现有的技术,因为它没有对人口统计学、连锁或显性做出任何假设。我们讨论了我们的方法如何为从新的角度研究自然选择铺平道路。