Folkhälsan Research Center, Helsinki, Finland and Medicum, University of Helsinki, FI-00290 Helsinki, Finland.
Klinik und Poliklinik für Neurologie, Sektion Neuromuskuläre Erkrankungen, Universitätsklinikum Bonn, D-53127 Bonn, Germany.
Hum Mol Genet. 2023 Oct 17;32(21):3029-3039. doi: 10.1093/hmg/ddad058.
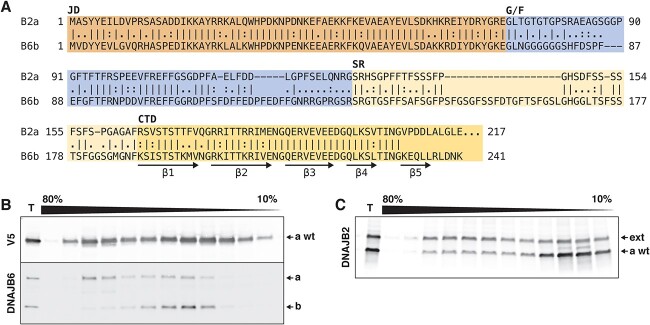
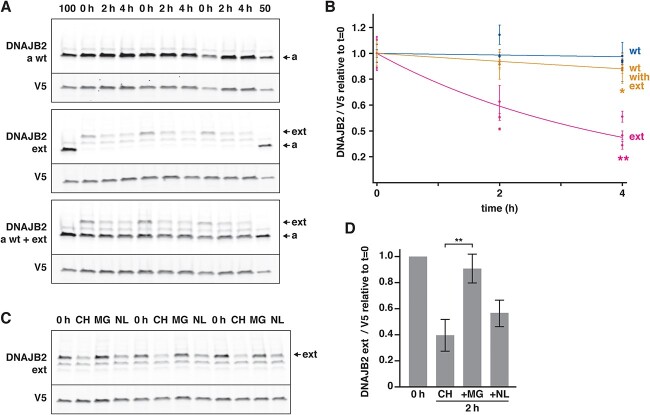
Recessive mutations in the DNAJB2 gene, encoding the J-domain co-chaperones DNAJB2a and DNAJB2b, have previously been reported as the genetic cause of progressive peripheral neuropathies, rarely involving pyramidal signs, parkinsonism and myopathy. We describe here a family with the first dominantly acting DNAJB2 mutation resulting in a late-onset neuromyopathy phenotype. The c.832 T > G p.(278Glyext83) mutation abolishes the stop codon of the DNAJB2a isoform resulting in a C-terminal extension of the protein, with no direct effect predicted on the DNAJB2b isoform of the protein. Analysis of the muscle biopsy showed reduction of both protein isoforms. In functional studies, the mutant protein mislocalized to the endoplasmic reticulum due to a transmembrane helix in the C-terminal extension. The mutant protein underwent rapid proteasomal degradation and also increased the turnover of co-expressed wild-type DNAJB2a, potentially explaining the reduced protein amount in the patient muscle tissue. In line with this dominant negative effect, both wild-type and mutant DNAJB2a were shown to form polydisperse oligomers.
先前已有研究报道,DNAJB2 基因(编码 J 结构域共伴侣 DNAJB2a 和 DNAJB2b)的隐性突变是进行性周围神经病的遗传原因,很少伴有锥体束征、帕金森病和肌病。我们在此描述了一个家族,该家族的首个显性作用的 DNAJB2 突变导致迟发性神经肌肉病表型。c.832T>G p.(278Glyext83) 突变导致 DNAJB2a 异构体的终止密码子缺失,从而导致蛋白质 C 末端延伸,对该蛋白的 DNAJB2b 异构体没有直接影响。肌肉活检分析显示两种蛋白异构体的减少。在功能研究中,由于 C 末端延伸中的跨膜螺旋,突变蛋白错误定位到内质网。突变蛋白迅速被蛋白酶体降解,并增加共表达的野生型 DNAJB2a 的周转率,这可能解释了患者肌肉组织中蛋白数量减少的原因。与这种显性负效应一致,野生型和突变型 DNAJB2a 均形成多分散的寡聚物。