Department of Clinical Neurosciences, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
Department of Diagnostics and Applied Technology, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
Eur J Neurol. 2022 Jul;29(7):2056-2065. doi: 10.1111/ene.15326. Epub 2022 Mar 23.
Mutations in DNAJB2 are associated with autosomal recessive hereditary motor neuropathies/ Charcot-Marie-Tooth disease type 2 (CMT2). We describe an Italian family with CMT2 due to a homozygous DNAJB2 mutation and provide insight into the pathomechanisms.
Patients with DNAJB2 mutations were characterized clinically, electrophysiologically and by means of skin biopsy. mRNA and protein levels were studied in lymphoblastoid cells (LCLs) from patients and controls.
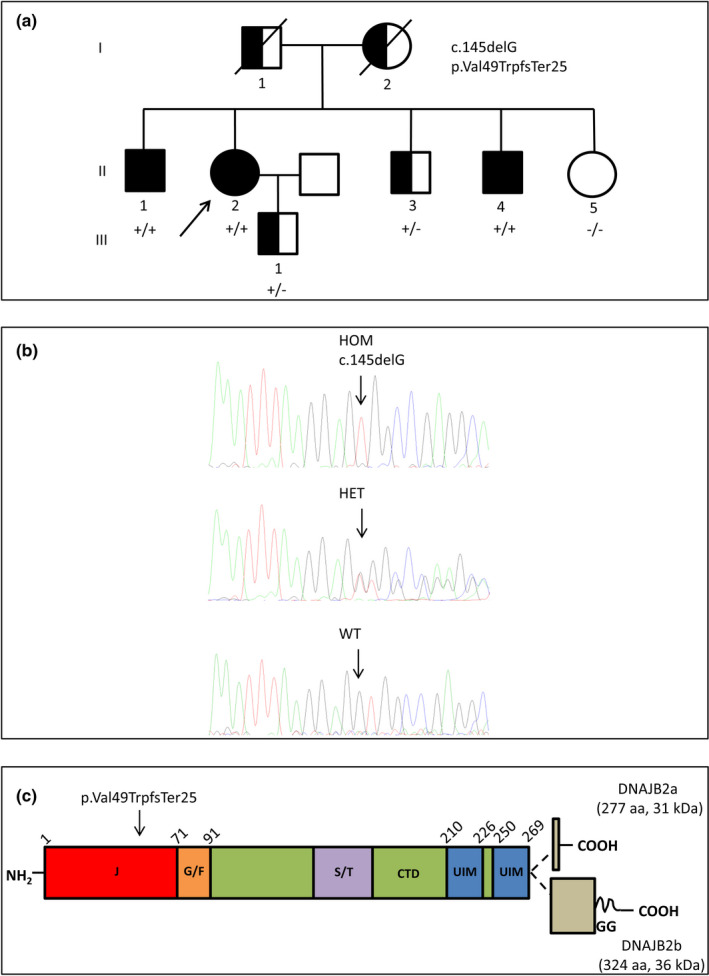
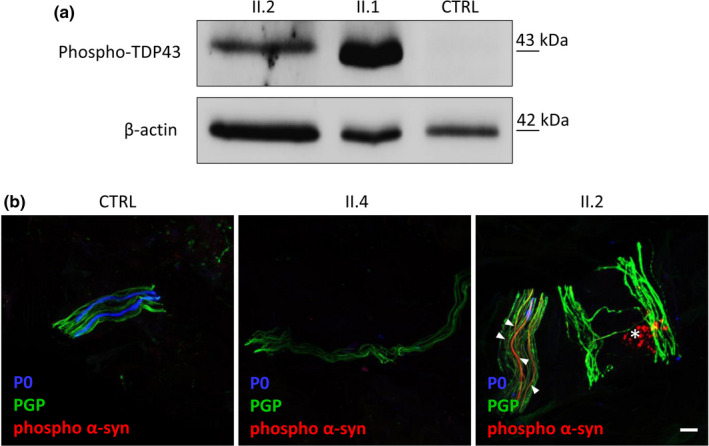
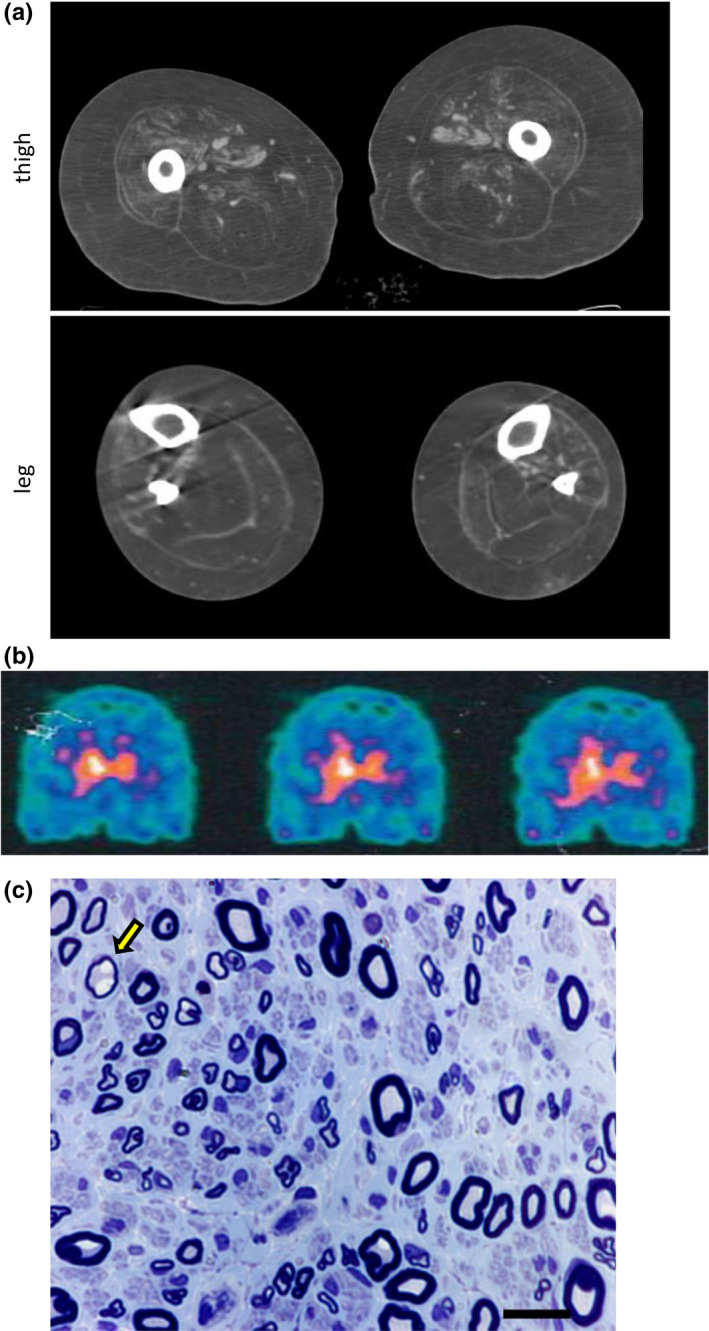
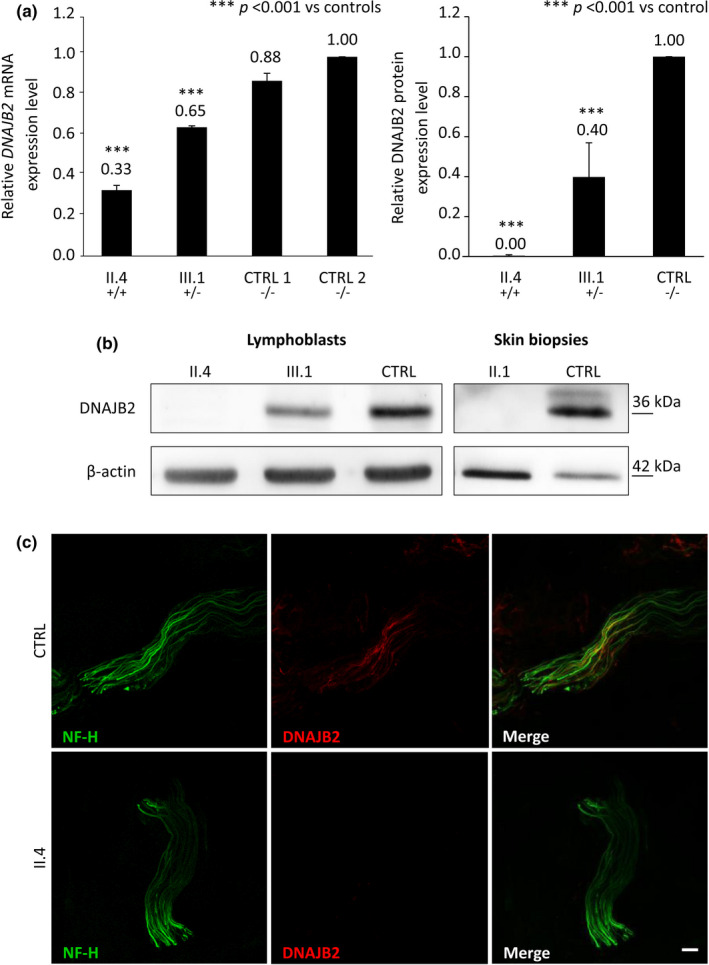
Three affected siblings were found to carry a homozygous DNAJB2 null mutation segregating with the disease. The disease manifested in the second to third decade of life. Clinical examination showed severe weakness of the thigh muscles and complete loss of movement in the foot and leg muscles. Sensation was reduced in the lower limbs. All patients had severe hearing loss and the proband also had Parkinson's disease (PD). Nerve conduction studies showed an axonal motor and sensory length-dependent polyneuropathy. DNAJB2 expression studies revealed reduced mRNA levels and the absence of the protein in the homozygous subject in both LCLs and skin biopsy. Interestingly, we detected phospho-alpha-synuclein deposits in the proband, as already seen in PD patients, and demonstrated TDP-43 accumulation in patients' skin.
Our results broaden the clinical spectrum of DNAJB2-related neuropathies and provide evidence that DNAJB2 mutations should be taken into account as another causative gene of CMT2 with hearing loss and parkinsonism. The mutation likely acts through a loss-of-function mechanism, leading to toxic protein aggregation such as TDP-43. The associated parkinsonism resembles the classic PD form with the addition of abnormal accumulation of phospho-alpha-synuclein.
DNAJB2 基因突变与常染色体隐性遗传性运动神经病/Charcot-Marie-Tooth 病 2 型(CMT2)相关。我们描述了一个意大利家族,该家族因 DNAJB2 突变纯合子导致 CMT2,并深入探讨了其发病机制。
对 DNAJB2 突变患者进行临床、电生理学和皮肤活检特征分析。对患者和对照的淋巴母细胞系(LCL)进行 mRNA 和蛋白水平研究。
发现 3 名受影响的兄弟姐妹携带纯合 DNAJB2 无义突变,与疾病共分离。疾病在第二至第三十年发病。临床检查显示大腿肌肉严重无力,足部和腿部肌肉完全丧失运动功能。下肢感觉减退。所有患者均有严重听力损失,先证者还患有帕金森病(PD)。神经传导研究显示轴索性运动和感觉神经长度依赖性多发性神经病。DNAJB2 表达研究显示,在 LCL 和皮肤活检中,纯合子患者的 mRNA 水平降低,且该蛋白缺失。有趣的是,我们在先证者中检测到磷酸化-α-突触核蛋白沉积,这在 PD 患者中已经观察到,并在患者皮肤中证实 TDP-43 积累。
我们的研究结果拓宽了 DNAJB2 相关神经病变的临床谱,并提供了证据表明 DNAJB2 突变应被视为另一个引起 CMT2 伴听力损失和帕金森病的致病基因。该突变可能通过丧失功能机制起作用,导致 TDP-43 等毒性蛋白聚集。相关帕金森病与经典 PD 形式相似,伴有异常磷酸化-α-突触核蛋白积累。