Systems Biology Initiative, School of Biotechnology and Biomolecular Sciences, The University of New South Wales, Randwick, NSW 2052, Australia.
School of Chemistry, University of Sydney, Sydney, NSW 2006, Australia.
Proc Natl Acad Sci U S A. 2023 Apr 25;120(17):e2219418120. doi: 10.1073/pnas.2219418120. Epub 2023 Apr 18.
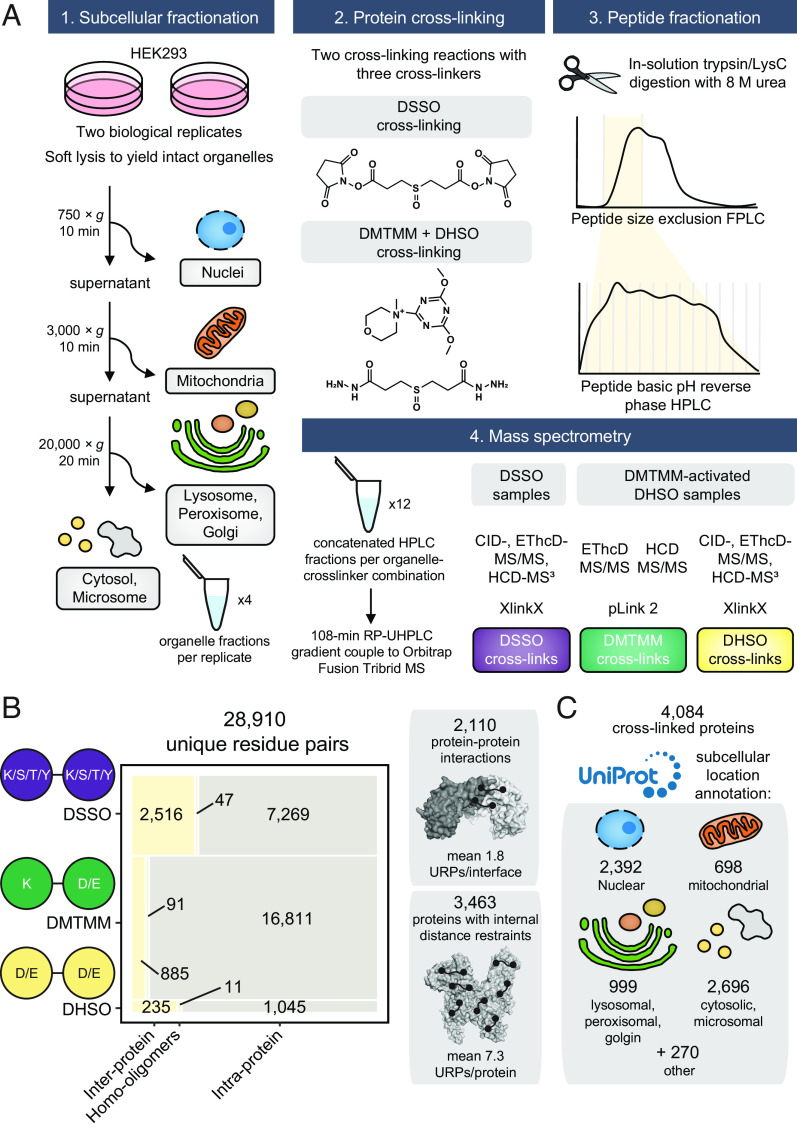
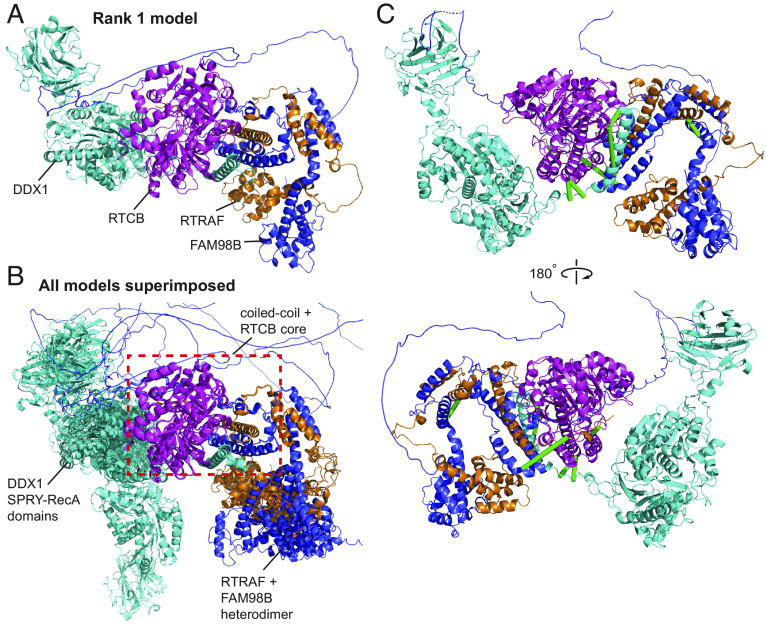
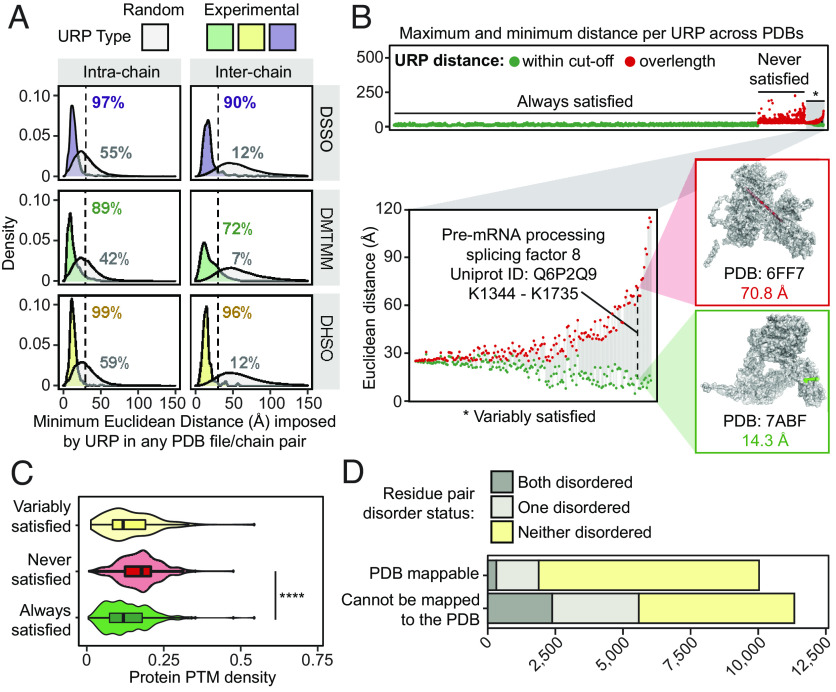
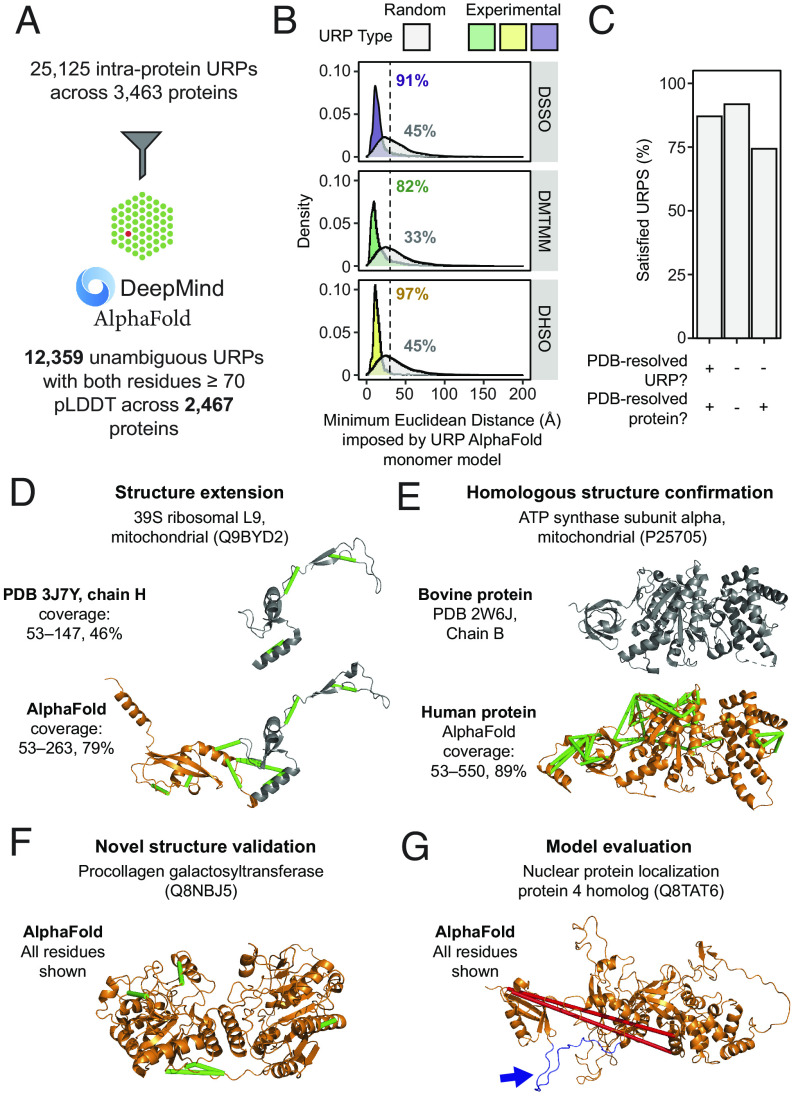
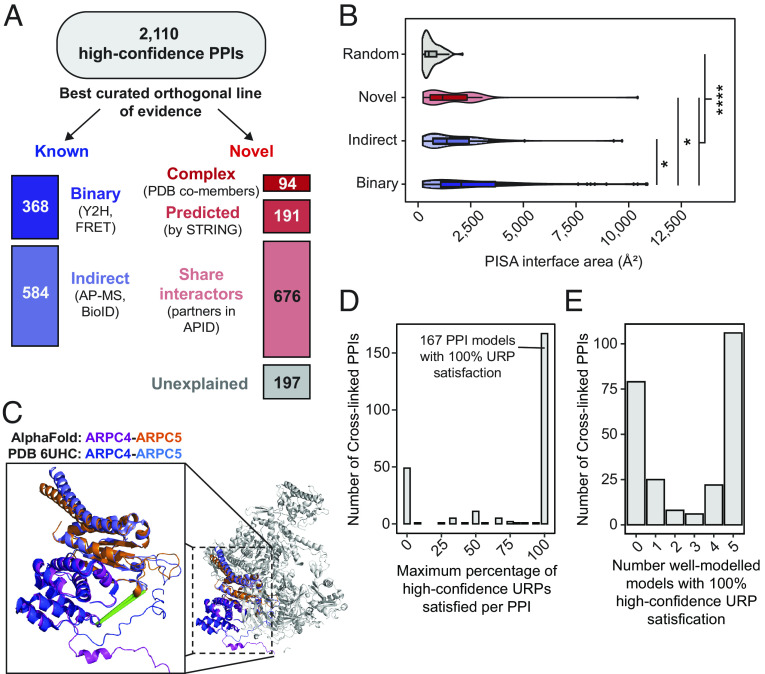
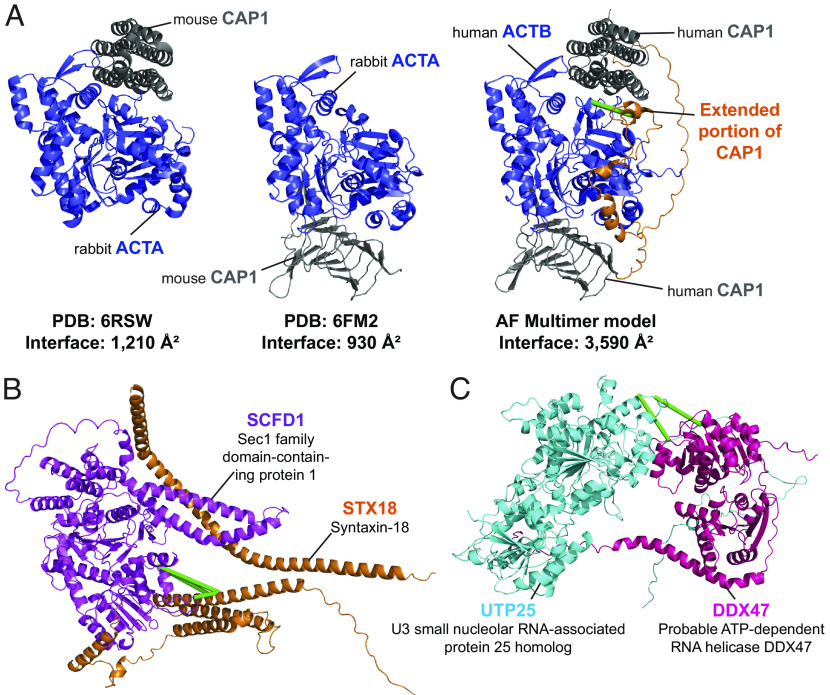
Significant recent advances in structural biology, particularly in the field of cryoelectron microscopy, have dramatically expanded our ability to create structural models of proteins and protein complexes. However, many proteins remain refractory to these approaches because of their low abundance, low stability, or-in the case of complexes-simply not having yet been analyzed. Here, we demonstrate the power of using cross-linking mass spectrometry (XL-MS) for the high-throughput experimental assessment of the structures of proteins and protein complexes. This included those produced by high-resolution but in vitro experimental data, as well as in silico predictions based on amino acid sequence alone. We present the largest XL-MS dataset to date, describing 28,910 unique residue pairs captured across 4,084 unique human proteins and 2,110 unique protein-protein interactions. We show that models of proteins and their complexes predicted by AlphaFold2, and inspired and corroborated by the XL-MS data, offer opportunities to deeply mine the structural proteome and interactome and reveal mechanisms underlying protein structure and function.
近年来,结构生物学,特别是冷冻电子显微镜领域取得了重大进展,极大地提高了我们构建蛋白质和蛋白质复合物结构模型的能力。然而,由于某些蛋白质丰度低、稳定性差,或者由于尚未进行分析,许多蛋白质仍然难以用这些方法进行研究。在这里,我们展示了使用交联质谱(XL-MS)进行高通量实验评估蛋白质和蛋白质复合物结构的强大功能。这包括使用高分辨率但体外实验数据以及仅基于氨基酸序列的计算预测产生的结构。我们提供了迄今为止最大的 XL-MS 数据集,描述了在 4084 个人类蛋白质和 2110 个独特的蛋白质-蛋白质相互作用中捕获的 28910 个独特残基对。我们表明,由 AlphaFold2 预测的蛋白质及其复合物模型,以及受到 XL-MS 数据启发和证实的模型,为深入挖掘结构蛋白质组和相互作用组以及揭示蛋白质结构和功能的机制提供了机会。